Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 24(3) - Setembro 2024
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Artigo Original
Perfil clínico de pacientes 46,xx com ambiguidade genital e diagnóstico de DDS: um estudo retrospectivo de um hospital público universitário
Clinical Profile of 46,XX Patients with Genital Ambiguity and DSD Diagnosis: A Retrospective Study from a Public University Hospital
Anna Candida Ximenes de Mendonça Sobreira; Paulo Ferrez Collett Solberg; Raquel Tavares Boy da Silva; Daniel Luis Schueftan Gilban; Clarice Borschiver de-Medeiros; Ana Paula Neves Bordallo; Claudia Braga Monteiro; Isabel Rey Madeira
DOI:10.31365/issn.2595-1769.v24i3p75-81
Hospital Universitário Pedro Ernesto - UERJ, Endocrinologia Pediátrica - Rio de Janeiro - RJ - Brasil
Endereço para correspondência:
Instituição: Faculdade de Ciências Médicas - UERJ - Rio de Janeiro - RJ - Brasil
Recebido em: 11/01/2024
Aprovado em: 23/07/2024
Resumo
Ambiguidade genital é um fenótipo de distúrbio do desenvolvimento sexual com incidência 1:4500, que constitui emergência clínica, exigindo rapidez no estabelecimento de etiologias graves e do sexo de criação.
OBJETIVO: Descrever o perfil dos pacientes 46,XX com ambiguidade genital.
MÉTODO: Estudo transversal retrospectivo de 17 prontuários.
RESULTADOS: O diagnóstico mais frequente foi hiperplasia adrenal congênita (11; 64,7%), seguido de DDS ovotesticular (4; 23,5%). O diagnóstico foi neonatal em 8 (47%). A idade média na primeira consulta foi de 55 meses. Houve discordância sexo social x genético em 2 (11,7%). História familiar positiva em 5 (29,4%). A genitália externa foi avaliada (escala de Prader) 3-5 em 15 (88,2%). Foi realizada intervenção cirúrgica em 10 (58,8%).
CONCLUSÃO: A hiperplasia adrenal congênita foi a etiologia mais comum, corroborando dados da literatura e deve ser o primeiro diagnóstico pensado na ambiguidade genital 46,XX. O diagnóstico neonatal possibilita manejo precoce. Apresentação tardia a serviço especializado leva a prejuízo da investigação e definição do sexo de criação, implicando morbimortalidade. Discordâncias entre sexo social e genético ressaltam a importância de avaliação multidisciplinar para evitar designações precipitadas. O alto grau de virilização da genitália externa pode justificar o número de cirurgias, embora cirurgias definitivas precoces sejam controversas. Repercussões do diagnóstico tardio e manejo inadequado impactam no relacionamento do indivíduo consigo e com a sociedade.
Palavras-chave: Desenvolvimento Sexual. Genitália. Cariótipo. Transtornos 46, XX do Desenvolvimento Sexual.
Abstract
Genital ambiguity is a phenotype of Disorders of Sex Development (DSD) with an incidence of 1:4500. This diagnosis constitutes a clinical and social emergency, requiring investigation to detect serious etiologies and gender determination.
OBJECTIVE: Describe the profile of patients with a karyotype of 46,XX, genital ambiguity and DSD diagnosis.
METHOD: Retrospective cross-sectional study analyzing 17 medical records.
RESULTS: The most frequent diagnosis was Congenital Adrenal Hyperplasia (CAH) in 11 cases (64.7%), followed by ovotesticular DSD in 4 cases (23.5%). Neonatal diagnosis was achieved in 8 cases (47%). The average age at the first consultation was 55 months. There was discordance between social and genetic gender in 2 cases (11.7%). Family history was positive in 5 cases (29.4%). External genitalia, assessed on the Prader scale, ranged from 3 to 5 in 15 cases (88.2%). Surgical intervention was performed in 10 cases (58.8%).
CONCLUSION: CAH, the most common etiology, aligns with existing literature and should be the primary consideration for 46,XX with genital ambiguity. Neonatal diagnosis enables early management; however, delayed presentation to specialized services may lead to morbidity, mortality, and hinder the investigation and determination of gender. Discrepancies between social and genetic gender underscore the importance of a multidisciplinary assessment to prevent premature designations. The high degree of virilization justifies the number of surgeries, although the controversy persists regarding early definitive procedures. Repercussions of late diagnosis and improper management impact the individuals' relationship with themselves and society.
Keywords: Disorders of Sex Development. Genitalia. Karyotype. 46, XX Disorders of Sex Development.
INTRODUÇÃO
As diferenças no desenvolvimento sexual (DDS) são condições congênitas nas quais o desenvolvimento dos sexos cromossômico, gonadal e anatômico são atípicos ou incongruentes entre si. A apresentação clínica das DDS é extremamente variável, sendo definida na presença de um dos seguintes critérios: ambiguidade genital evidente; genitália aparentemente feminina com clitoromegalia, fusão labioescrotal posterior ou massa inguinal/labioescrotal; genitália aparentemente masculina com criptorquidia bilateral, micropênis, hipospadia perineal isolada ou hipospádia leve com criptorquidia unilateral; história familiar de DDS; discordância entre fenótipo e cariótipo pré-natal.1
A ambiguidade genital (AG) consiste em um fenótipo de DDS e pode ser definida sempre que o médico apresente dificuldade para atribuir o sexo genital externo a determinada criança. Não há estimativas precisas sobre a incidência de ambiguidade genital ao nascimento, podendo variar de 1:4.500-5.500 nascidos vivos, embora apenas uma parcela seja um grande desafio para atribuição de sexo. A incidência de indivíduos com diagnóstico de DDS 46,XX está estimada em 1:14.000-15.000 nascidos vivos.2,3,4
Na primeira avaliação de um indivíduo com DDS, seja um recém-nascido, uma criança, um adolescente ou adulto, o primeiro objetivo é chegar ao diagnóstico sindrômico e, sempre que possível, a um diagnóstico etiológico preciso. Em geral, a suspeita clínica inicial cabe ao pediatra assistente na primeira avaliação ao nascimento ou em consulta de rotina. No entanto, não é incomum que a suspeita de que há algo errado com a genitália da criança seja feita a partir de um membro familiar.2,5
Trata-se de uma situação complexa não só por envolver condições que trazem risco à vida do recém-nascido, como a hiperplasia adrenal congênita (HAC) e quadros malformativos, mas também pelos efeitos psicossociais relacionados à indefinição do sexo social. Sua abordagem requer, portanto, ações multidisciplinares complexas, ágeis e eficazes iniciadas logo após a suspeita clínica 2,4,6
A HAC é uma doença genética autossômica recessiva caracterizada por um erro inato na síntese dos esteroides adrenais. Na HAC clássica (1:14.199), a deficiência da 21-hidroxilase é a responsável por mais de 90% dos casos e se apresenta em dois fenótipos principais. Na forma virilizante simples, acarreta aumento de andrógenos e provoca AG em graus variados nas meninas; enquanto a forma perdedora de sal (PS), além da AG, compromete a produção de aldosterona e pode gerar uma crise de insuficiência adrenal por deficiência acentuada na reabsorção de sódio, resultando em hiponatremia potencialmente letal nas primeiras semanas de vida.2,7,8
Considerando a gravidade potencial desta condição, o rastreamento neonatal, através da implementação de programas como o Teste de Triagem Neonatal (Teste do Pezinho) adotado no Brasil, tem se revelado fundamental na detecção precoce da HAC. Essa prática sistemática facilita a instauração tempestiva de medidas terapêuticas, contribuindo de forma significativa para a mitigação da morbimortalidade associada à patologia.5,9
O grande desafio frente ao paciente com DDS, principalmente crianças com AG, é chegar a um diagnóstico etiológico preciso. Nesse processo, anamnese e exame físico criteriosos do paciente, bem como exames de imagem podem direcionar a investigação diagnóstica.2,5
Na anamnese, devem ser avaliados os antecedentes gestacionais, com especial atenção ao uso de medicamentos e a sinais de virilização materna na gestação, além de história de baixo peso ao nascimento. Importante também perguntar ativamente sobre antecedentes familiares, como consanguinidade entre os pais, casos semelhantes na família, história familiar de atraso ou avanço puberal, infertilidade, hipertensão arterial na infância ou mortes inexplicadas nos primeiros meses de vida. 2,5,8
A genitália externa deve ser avaliada determinando-se o grau de virilização, com análise de: tamanho do falo; posição do meato uretral; presença de intróito vaginal ou abertura de seio urogenital; grau de fusão, simetria, pigmentação e enrugamento das saliências labioescrotais; presença de massas inguinais; assim como a localização e tamanho das gônadas. Existem sistemas para avaliar a gravidade da ambiguidade genital como as escalas de Prader (Figura 1), elaborada em 1954 inicialmente para os casos de meninas com diagnóstico de HAC, que apresentam graus progressivos de AG, estendendo-se desde a aparência predominantemente feminina à predominantemente masculina.2,5,8, 10
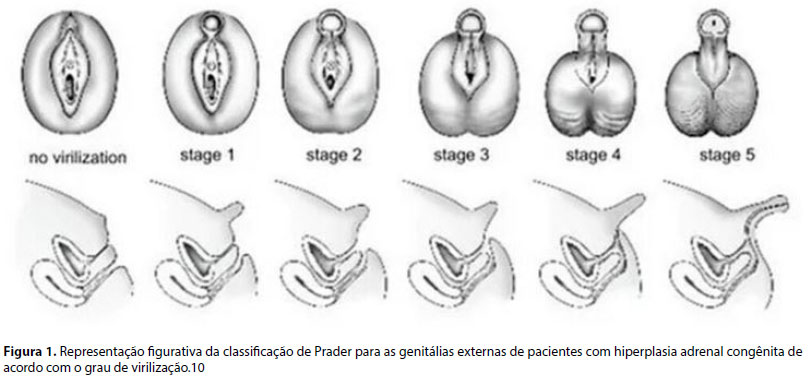
As gônadas localizadas em região inguinal podem ser detectadas pela palpação e pela ultrassonografia (US), sendo que este exame de imagem também pode identificar gônadas intra-abdominais, útero e anormalidades adrenais. No entanto, o exame tem capacidade limitada de localizar gônadas intra-abdominais e discriminar ovário de testículo, além de nem sempre conseguir visualizar as gônadas e o útero, podendo ser necessária complementação com outras modalidades de exame de imagem, como a ressonância nuclear magnética (RM).8, 11
Os critérios para determinar o sexo de criação são: sexo genético/molecular, o fenotípico (sexo gonadal e potencialidade hormonal e gametogênica mais genitália interna e externa), e sexo psicológico e cerebral. Portanto, deve ser estabelecido somente após cuidadosa avaliação da equipe e o mais breve possível, visto a situação de estresse vivenciada pela família. Desse diagnóstico depende não só a definição do sexo, mas também todos os procedimentos terapêuticos subsequentes e ainda o aconselhamento genético da família.2,4,8,11
O objetivo do estudo é descrever a análise dos pacientes com cariótipo 46,XX com AG e diagnóstico de DDS que são acompanhados no serviço de endocrinologia pediátrica de um hospital universitário de referência da rede pública.
MÉTODO
Foi realizado estudo de observação transversal retrospectivo com base na análise de prontuários de indivíduos encaminhados ao Setor de Endocrinologia Pediátrica de um hospital público universitário do Estado do Rio de Janeiro. Este é um ambulatório de ensino, que faz parte da rede do Sistema Único de Saúde (SUS), onde se presta atenção primária, secundária e terciária às crianças, referidas do Ambulatório de Pediatria Geral do mesmo hospital e da rede pública, do nascimento até 17 anos de idade.
Este estudo foi aprovado pelo Comitê de Ética e Pesquisa, onde está cadastrado com o número CAAE 34116420.9.0000.5259.
Foram incluídos crianças e adolescentes, do nascimento a 17 anos, que apresentavam diagnóstico de DDS 46,XX com apresentação fenotípica de ambiguidade genital na primeira consulta, conforme critérios estabelecidos no Consenso DDS 2006. Após a aplicação desses critérios, foram selecionados prontuários de 17 indivíduos. Estes foram analisados em relação a idade na primeira consulta, idade do registro civil, história gestacional, história familiar, presença de consanguinidade, exames de imagem realizados, cariótipo, sexo social, concordância com sexo genético e mudança de sexo no registro civil. Em relação ao exame físico na primeira consulta, foram analisadas descrição da genitália, aplicação da escala de Prader, presença de gônadas palpáveis, lateralidade destas dismorfias ou malformações congênitas. Por fim, foram categorizados os diagnósticos estabelecidos e os tratamentos propostos.1,10
Foi valorizada, na história gestacional, qualquer intercorrência ou uso de medicações durante a gestação. Foi considerada história familiar significativa a presença de parentes de primeiro grau com diagnóstico estabelecido de DDS ou história de ambiguidade genital. A consanguinidade foi definida como qualquer grau de parentesco entre os genitores.
Os exames de imagens realizados constituíram-se de US de abdome, pelve e eminências labioescrotais, além de RM de abdome e pelve, conforme necessário para investigação de cada caso. Os cariótipos banda G, obtidos de amostras de sangue periférico, foram realizados em diferentes laboratórios, sendo a maioria das amostras processadas e analisadas no Laboratório de Citogenética da instituição, seguindo protocolo padrão.12
Foi definido sexo social como o sexo estabelecido no registro civil do paciente. Para classificação do grau de virilização da genitália, foi utilizada a escala de Prader.10
Os dados coletados foram arquivados em planilhas Sheets acessadas e armazenadas no Google Drive e analisados no próprio site.
RESULTADOS
Foram analisados 17 prontuários de pacientes com cariótipo 46,XX atendidos no serviço entre os anos de 2010 a 2023 e que possuíam na primeira consulta o diagnóstico sindrômico de ambiguidade genital. Em relação ao cariótipo, todos os pacientes apresentaram o cariótipo 46,XX, sendo este um critério de inclusão indispensável para o estudo.
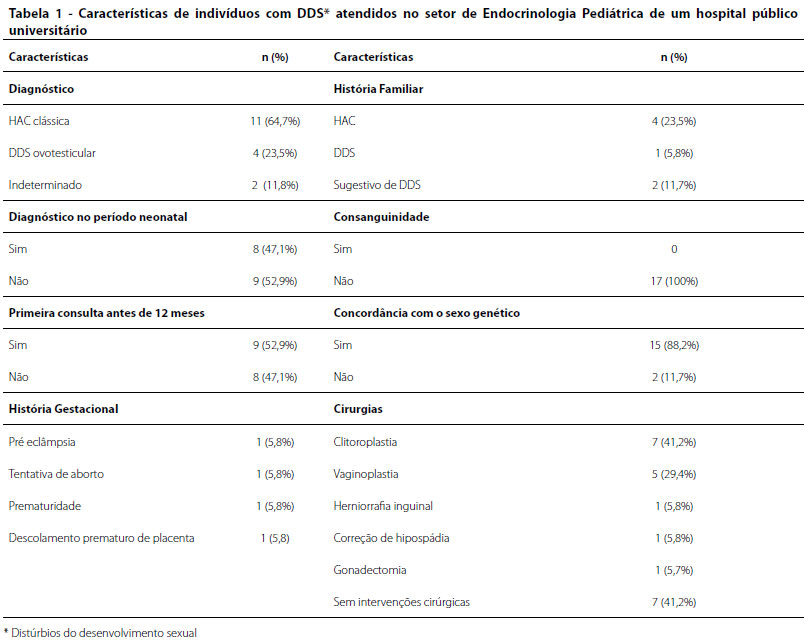
Na Tabela 1, estão resumidas algumas características observadas nos indivíduos com diagnóstico de DDS com cariótipo 46,XX atendidos no Setor de Endocrinologia Pediátrica. O diagnóstico mais frequente foi HAC clássica em 11 casos (64,7%), seguido de DDS ovotesticular em 4 (23,5%). Nota-se que não foi possível concluir a avaliação diagnóstica em dois casos (11,8%), devido à indisponibilidade de exames moleculares específicos ou abandono de acompanhamento. O diagnóstico no período neonatal, até 30 dias de vida, foi alcançado em 8 (47%).
A média de idade na primeira consulta no ambulatório de endocrinologia foi de 55 meses, sendo observado que somente nove indivíduos (52,9%) realizaram a primeira consulta nos primeiros 12 meses de vida e, destes, apenas 3 (17,6%) no período neonatal. De forma inversa, sete indivíduos já tinham quatro anos ou mais na primeira consulta.
Na época de admissão no serviço, 12 pacientes (70,6%) já apresentavam sexo social estabelecido, apesar do quadro de ambiguidade genital. A média de idade do registro civil foi de 0,9 meses. O sexo social adotado inicialmente foi feminino em 14 (82,3%) e masculino em 3 (17,6%), sendo um paciente alterado para feminino após o diagnóstico. Houve discordância entre sexo social e genético em dois casos (11,7%).
A história familiar foi positiva em parentes de primeiro grau em 5 (29,4%), tais como HAC e DDS. Outros 2 (11,8%) apresentaram história familiar sugestiva de outros casos DDS na família, com relato de outros parentes com micropênis, cirurgia genital e infertilidade. Nenhum apresentava história de consanguinidade entre os pais. A história gestacional foi relevante em 4 (23,4), sendo as ocorrências pré-eclâmpsia, descolamento prematuro de placenta, tentativa falha de aborto e prematuridade.
No exame físico descrito na apresentação do paciente ao serviço, observaram-se falo maior que 2 cm em 15 pacientes (88,2%) e eminências labioescrotais completamente fundidas em 7 (41,2%). O estadiamento de Prader aplicado na genitália externa foi avaliada em grau 3 a 5 em 15 (88,2%), sendo grau 5 em um paciente, grau 4 em seis, grau 3 em oito. Em dois indivíduos (11,7%) não foi realizada adequada caracterização da genitália para que fossem classificados.
Em relação ao tratamento, 10 pacientes (58,8%) realizaram alguma intervenção cirúrgica, sendo a cistoplastia redutora a mais frequente, realizada em seis pacientes (35,3%).
DISCUSSÃO
Os distúrbios que afetam a determinação e a diferenciação do sexo trazem consigo graves implicações médicas, psicológicas e sociais. Portanto, é necessária uma investigação complexa, ágil e eficaz, que deve ser abordada de maneira interdisciplinar com pediatra, endocrinologista, cirurgião, geneticista clínico, psicólogo, assistente social, entre outros.
Uma primeira abordagem para esclarecer a etiologia é posicionar a ambiguidade genital como um fenótipo do grupo dos DDS. Estas são condições congênitas nas quais há discordância entre os achados cromossômicos, gonadais e anatômicos da genitália.4,13
A identificação e investigação desses casos nesse período é fundamental para a detecção de casos potencialmente letais, como a HAC em sua forma perdedora de sal e síndromes malformativas. Na casuística descrita, diferentemente do preconizado, apenas 73,3% dos pacientes tiveram seu diagnóstico ainda no período neonatal. Embora tenha constituído a maioria dos casos, o fato de mais de um quarto dos casos terem tido o diagnóstico depois do período neonatal é preocupante.
O impacto psicossocial da indefinição de sexo na ambiguidade genital geralmente leva as famílias a buscarem atendimento o mais precocemente possível. Contudo, medo e ansiedade são situações de destaque entre os pais de um recém-nascido com ambiguidade genital. A negação e fuga são formas comuns de enfrentar essas situações. Aventa-se a possibilidade de que, em alguns casos, a dificuldade em lidar com o diagnóstico tenha contribuído para o atraso da primeira avaliação em idade adequada.2,8,14
Outros aspectos que contribuem para o diagnóstico tardio incluem carência de conhecimento médico a respeito da genitália normal e falta de entendimento sobre as situações de variação da normalidade, ausência de pediatra na sala de parto e nas equipes de Saúde da Família, o que impossibilita o exame físico detalhado, bem como a dificuldade no encaminhamento na coordenação do cuidado. Dessa forma, evidencia-se a necessidade de treinamento e capacitação dos profissionais, a fim de incentivar a rápida identificação e o encaminhamento dos casos suspeitos ou que gerem dúvidas para uma avaliação dentro de um serviço especializado.2,5,8,11
Enfatiza-se a importância do reconhecimento e valorização dos sinais de ambiguidade genital pelos profissionais que avaliam rotineiramente a criança na maternidade ou na atenção primária à saúde. Neste sentido, a necessidade de educação permanente paras pediatras / neonatologistas em relação à ambiguidade genital é outro aspecto crítico.2,8,15
A idade média de apresentação ao serviço foi de 54 meses, sendo inferior à observada em outras casuísticas semelhantes, como a realizada na Tailândia, em que a idade média de apresentação foi de 80 meses. Nesse mesmo trabalho, foi observado que oito indivíduos foram avaliados antes de um ano de idade, representando um pouco mais da metade da amostra. Outra casuística descrita por outro centro de referência brasileiro relata que 50,6% dos casos de ambiguidade genital foram encaminhados no primeiro ano de vida, mostrando que mesmo em outros centros ainda existe atraso nesse tipo de encaminhamento.15
O diagnóstico mais frequente foi a HAC clássica, o que corrobora os dados da literatura, de que a forma clássica por deficiência da enzima 21-hidroxilase é responsável por mais de 90% dos casos, principal etiologia em casos de ambiguidade genital e das DDS 46,XX. No entanto, ressalta-se que essa não é a única forma de apresentação da afecção, uma vez que depende de qual atividade enzimática está deficiente e do grau dessa deficiência.2,7,8
A segunda etiologia mais frequente foi DDS ovariotesticular, cujo diagnóstico é obrigatoriamente histopatológico e depende da presença concomitante de tecido ovariano e testicular em um mesmo indivíduo.2,8
Em um caso não foi possível fechar o diagnóstico, sendo ele indeterminado por não fechar os critérios laboratoriais para HAC e devido à ausência de exames citogenéticos mais específicos. Apesar de estar em um centro de referência, ainda há a dificuldade de acesso a tais exames.
É fundamental o diagnóstico antes do estabelecimento da identidade sexual social e psicológica, uma vez que o diagnóstico preciso não só permitirá estabelecer a correta definição de sexo, como também o prognóstico individual, a conduta médica e cirúrgica; permitirá, ainda, lançar mão de subsídios para o aconselhamento genético da família ou mesmo do próprio indivíduo, nos casos em que a fertilidade é preservada.2,6,8
Apesar da idade mais precoce de apresentação observada em nosso serviço ao se comparar com outras casuísticas, o ideal é que a avaliação de um recém-nascido com genitália ambígua seja cuidadosamente iniciada ainda no período neonatal, e muitas vezes ainda no pré-natal. A relevância imediata está na identificação de etiologias que podem colocar a vida da criança em risco e, no longo prazo, por uma definição de sexo mal-resolvida, que pode acarretar prejuízos irreparáveis ao bem-estar psicossocial do paciente.
Em relação ao registro civil, nota-se que nove pacientes foram registradas logo ao nascer, mesmo com a presença da genitália ambígua e ausência de diagnóstico. Entretanto, a orientação é de que a criança não deva ser registrada enquanto não se tenha esclarecido claramente o diagnóstico, ajudando a evitar algumas cicatrizes que o registro inadequado possa acarretar.6
Recomenda-se registro feminino em indivíduos 46,XX com HAC, dos quais mais de 90% têm identidade de gênero feminino. Entretanto, na presente casuística, um indivíduo foi registrado inicialmente como menino e posteriormente modificado para menina após o diagnóstico de HAC.2,8
Observou-se que os 15 indivíduos se apresentavam com importante ambiguidade genital, com graus de Prader entre 3 e 5. Entretanto, talvez meninas com graus menores de virilização estejam sendo subdiagnosticadas.10
Dentre os princípios éticos sobre o manejo do DDS, estão os de procurar minimizar os riscos físicos e psicossocial; preservar o potencial de fertilidade; reconhecer o direito do indivíduo de participar de decisões que irão afetá-lo naquele momento e no futuro; evitar procedimentos irreversíveis que sejam desnecessários do ponto de vista médico até que o indivíduo tenha capacidade de consentir; proporcionar suporte psicossocial e psicossexual; fornecer aos pacientes informações médicas completas de forma apropriada à idade, ao estágio do desenvolvimento e à capacidade cognitiva. 4,6,8
A irreversibilidade que caracteriza as propostas cirúrgicas de intervenção de ambiguidade genital nos DDS, somada à incapacidade legal e cognitiva dos indivíduos em idade tenra para oferecer consentimento, tem sido objeto de intenso debate, juntamente com considerações sobre aspectos técnicos, possíveis complicações e resultados no longo prazo. Assim, cabe a terceiros, pais ou responsáveis informados pela equipe multidisciplinar, consentirem. Entretanto, a decisão dos pais em solicitar a cirurgia precoce reduz as opções da criança no futuro, sem que esta possa participar da decisão. ,4,8
CONCLUSÃO
A HAC foi a etiologia mais comum de DDS 46,XX, o que corrobora os dados descritos na literatura e evidencia que deve ser o primeiro diagnóstico a ser pensado em ambiguidade genital, quando o cariótipo é este, por sua frequência e morbimortalidade. Preconiza-se o diagnóstico idealmente no período neonatal para detecção e manejo precoce de casos potencialmente letais, o que foi alcançado em 53,3% dos casos. Entretanto, a primeira consulta ao serviço é tardia e pode prejudicar o seguimento da investigação e definição do sexo de criação o mais breve possível por equipe especializada.
Foi adotado sexo feminino em 86,6% em concordância com a literatura. Disparidades entre sexo social e genético ressaltam a importância de avaliação criteriosa por equipe multidisciplinar experiente, a fim de evitar uma designação precipitada. O alto grau de virilização da genitália externa pode justificar o número de genitoplastias, embora ainda haja controvérsias na literatura acerca de cirurgias definitivas precoces.
Repercussões do diagnóstico tardio e manejo inadequado podem gerar impactos irreparáveis no relacionamento do indivíduo consigo e com a sociedade. Assim, é fundamental que o pediatra esteja atento a esta possibilidade diagnóstica e realize o adequado encaminhamento desse indivíduo.
REFERÊNCIAS
1. Lee PA, Houk CP, Ahmed SF, Hughes IA. International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. Pediatrics. 2006;118:e488-500.
2. Maciel-Guerra AT, Guerra-Júnior G. Menino ou menina? Os distúrbios da diferenciação do sexo: volume 1. 3ª ed. Curitiba: Appris; 2019. 343 p.
3. Hamerton JL, Canning N, Ray M, Smith S. A cytogenetic survey of 14,069 newborn infants. I. Incidence of chromosome abnormalities. Clin Genet. 1975;8:223-43.
4. Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, et al. Global DSD Update Consortium. Global disorders of sex development update since 2006: perceptions, approach and care. Horm Res Paediatr. 2016;85:158-80.
5. Guerra-Júnior G, Maciel-Guerra AT. O pediatra frente a uma criança com ambiguidade genital. J Pediatr (Rio J). 2007;83(5 Suppl):S184-91.
6. Oliveira ACG de A. Os corpos refeitos: a intersexualidade, a prática médica e o direito à saúde. Rev Gênero Sexualidade Direito. 2015;2:1-25.
7. Speiser PW, Arlt W, Auchus RJ. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2018;103:4043-88.
8. Maciel-Guerra AT, Guerra-Júnior G. Menino ou menina? Os distúrbios da diferenciação do sexo: volume 2. 3ª ed. Curitiba: Appris; 2019. 321 p.
9. Brasil. Ministério da Saúde. Secretaria de Atenção à Saúde. Departamento de Atenção Especializada. Manual técnico do teste do pezinho. Brasília: Ministério da Saúde; 2016.
10. Prader A. Der genitalbefund beim pseudohermaproditus feminus des kongenitalen andrenogenitalen syndromes. Helv Paediatr Acta. 1954;9:231-48.
11. Wherrett DK. Approach to the Infant with a suspected disorder of sex development. Pediatr Clin North Am. 2015;62:983-99.
12. Seabright M. A rapid banding technique for human chromosomes. Lancet 1971;2:971-2.
13. Hughes IA. Consensus statement on management of intersex disorders. J Pediatr Urol. 2006;2:148-62.
14. De Paula GB. 408 cases of genital ambiguity followed by single multidisciplinary team during 23 years: etiologic diagnosis and sex of rearing. Int J Endocrinol. 2016;2016:496-574.
15. Beck MSE, Germano CW, Barros BA, Andrade JGR, Guaragna-Filho G, Paula GB, et al. Why pediatricians need to know the disorders of sex development: experience of 709 cases in a specialized service. J Pediatr (Rio J). 2020;96:607-13.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()