Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 10(1) - Maio 2009
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Dificuldades no diagnostico e manejo da acidemia metilmalônica - relato de caso sugestivo
Difficulties in diagnosis and treatment of methylmalonic aciduria - suggestive case report
Raphaella de Oliveira Cunha Trindade 1; Raquel Siqueira Leonel De Paula 2; Carlos Alberto Bhering 3; Isaias Soares De Paiva 4
1. Residencia Medica (Medico Residente em Pediatria).
2. Medico residente em pediatria (medico).
3. Doutor (coordenador uti neonatal).
4. Doutor (geneticista).
Endereço para correspondência:
Av Domingos Mariano 339 apto 504
Centro
Barra Mansa
cep 27345-310
Resumo
INTRODUÇÃO: A acidemia metilmalônica (AMM) é um erro inato do metabolismo (EIM) dos ácidos orgânicos. Existem duas formas clínicas : uma neonatal e outra tardia, a maioria com apresentação súbita no período neonatal. Os afetados apresentam sintomas graves, que muitas vezes os levam ao óbito, e são freqüentemente confundidos com septicemia, pois ambas as situações apresentam acidose e encefalopatia aguda.OBJETIVO: Relatar um recém-nascido com quadro sugestivo de AMM que evoluiu para o óbito por dificuldades e demora no diagnóstico e tratamento, visando a divulgação do conhecimento deste EIM.
MATERIAIS E MÉTODOS: Estudo descritivo constando de relato de caso.
RESULTADOS: KSC, 22 dias de vida, foi trazido ao pronto-socorro com história de intolerância alimentar desde o nascimento. O RN estava desnutrido grau III e desidratado e foi internado na UTI neonatal. Considerado sepse e iniciado esquema empírico de antibioticoterapia. Após persistência do quadro, foi iniciado screening para EIM que mostrou aumento moderado de C3. Os ciclos de metronidazol, a suplementação de L-carnitina e megadoses de vitaminas foram feitos, como recomenda o protocolo. Com a dificuldade em se obter a fórmula láctea recomendada (XMTVI analog), foi tentada associação de TCM e leite 80056, sem sucesso. O início da referida fórmula láctea deu-se no 87º dia de internação, dois dias antes do óbito do neonato.
CONSIDERAÇÕES FINAIS: Ressaltamos a necessidade do conhecimento da AMM, pois o prognóstico do paciente está relacionado ao tempo para instituir o tratamento adequado. As dificuldades em seguir o protocolo diagnóstico e terapêutico contribuem para um pior desfecho.
Palavras-chave: Acidemia Metilmalônica, Erro Inato do Metabolismo, Acidemias, Diagnostico, neonato
Abstract
INTRODUCTION: Methylmalonic aciduria (MMA) is an inborn error of metabolism (IEM) of organic acids. We can find two clinical forms, the first, in neonatal period and the other with late onset. Most of cases show up in neonatal period. These patients are seriously injured and most of them have fatal outcome and the symptoms are frequently confused with sepsis, since both of situations present acidosis and acute encephalopathy.OBJECTIVE: To relate a newborn with a suggested case of MMA who had a fatal outcome due to difficulties and delay in diagnosis and treatment, emphasizing the importance of knowing this disease.
MATERIALS AND METHODS: Descriptive study consisting of case report.
RESULTS: KSC, 22 days old, was brought to emergency with past of food intolerance since his birth.. The newborn was malnourished and dehydrated and was admitted to the Neonatal Intensive Care Unity. Sepsis was considered and empirical antibiotic therapy was introduced. A screening for IEM was started and has shown a moderate increase of C3. Metronidazole cycles, supplementation of L-carnitin and large doses of vitamins were done according to the recommended protocol for MMA's treatment. Because of the difficulty to obtain the recommended milt formula (XMTV analog®), another association was tried and failed. The referred milt formula was introduced at the 87º day placed in hospital, two days before newborn's death.
FINAL CONSIDERATIONS: We emphasize the need of knowing MMA, because the prognosis is related to the early diagnosis and appropriate treatment. Difficulties to follow diagnosis and treatment protocols contribute to a worse outcome.
Keywords: Methylmalonic Aciduria, Inborn Error Of Metabolism, Acidurias, Diagnosis, Newborn
1 - INTRODUÇÃO
A acidemia metilmalônica (AMM) é um dos mais freqüentes erros inatos do metabolismo (EIM) dos ácidos orgânicos. O defeito bioquímico se localiza no metabolismo do propionato, na etapa de conversão de ácido metilmalônico a ácido succínico. Existem dois tipos de alterações básicas: o defeito na apoenzima (metilmalonil-mutase) e o defeito de coenzimas (metil B-12 e 5-desoxiadenosil-B12)1[i]. A maioria dos pacientes tem um defeito na conversão de metilmalonil CoA a succinil CoA pelo defeito de apoenzima (metilmalonil-mutase). Esse grupo é o mais comum e não responde a administração de vitamina B12. No outro grupo, com defeito de coenzimas, a vitamina B12 (1mg/dia) reduz significativamente a excreção do ácido metilmalônico2[ii] 3[iii] 4[iv] 5[v] 6[vi] 7[vii].
Clinicamente se podem diferenciar duas formas, uma neonatal e outra de início tardio1, sendo a maioria com apresentação súbita no período neonatal8[viii]. Em ambas as formas, se destacam a existência de cetoacidose, dificuldade de desenvolvimento e hipotonia muscular1. A AMM se apresenta com crises de descompensação que são precipitadas pela ingesta de proteínas e de vários aminoácidos, principalmente leucina. As manifestações neurológicas são hipotonia e convulsões mioclônicas que podem levar ao coma2 3 4 9[ix] 10[x]. Os afetados apresentam sintomas graves, que muitas vezes os levam ao óbito, e são freqüentemente confundidos com septicemia, visto que ambas situações se caracterizam por acidose e encefalopatia aguda8. Embora rara, a prevalência da AMM é de 1 em 48000 na população geral11[xi].
É essencial que o pediatra se familiarize com a apresentação clínica dessas desordens, com uma investigação laboratorial mais adequada, com um melhor manejo na estabilização de pacientes criticamente doentes, e com a identificação daquelas crianças que poderiam se beneficiar de investigação e tratamento específicos12[xii].
O objetivo deste estudo é relatar um caso de um recém-nascido com quadro sugestivo de AMM que evoluiu para o óbito por dificuldades e demora no diagnóstico e tratamento, visando a divulgação do conhecimento deste EIM.
2 - MATERIAIS E MÉTODOS
Estudo descritivo constando de relato de caso. Os dados foram obtidos através de revisão de prontuário do paciente internado na Unidade de Terapia Intensiva Neonatal da Santa Casa de Misericórdia de Barra Mansa - RJ. Foi realizada também entrevista com os médicos assistentes, além de pesquisa na Internet em base de dados da literatura médica (Bireme, PubMed e Scielo) utilizando os termos "methylmalonic aciduria".
3 - RESULTADOS
KSC, 22 dias de vida, procedente de Barra Mansa, filho de JSC, 20 anos, dona de casa, GII PI AI, tipo sanguíneo O positivo. Pré-natal completo (10 consultas). Mãe referiu perda de líquido a partir do sétimo mês de gestação e oligodramnia. Nasceu de parto cesáreo, no termo, na 38ª; semana de gestação, sem evidências de asfixia. Pesou 2.990 gramas, mediu 46 centímetros, sendo classificado como adequado para a idade gestacional (AIG), tipo sanguíneo B positivo e de cor negra. Recebeu alta da maternidade com 48 horas de vida com aleitamento materno exclusivo.
O neonato é filho de pais jovens, não consangüíneos, aparentemente saudáveis, sem história de óbitos neonatais precoces anteriormente. Nega casos semelhantes em grupo familiar próximo.
Aos 22 dias de vida, o recém-nascido (RN) foi trazido ao pronto-socorro do hospital pela primeira vez. A mãe relatava história de intolerância alimentar desde o nascimento, com episódios de vômitos constantes e evacuações amareladas e amolecidas. Os vômitos eram pós-prandiais e a freqüência das evacuações aumentada. A mãe relatava também ter mudado o tipo de leite várias vezes sem sucesso.
No 22º dia de vida, o RN mostrava desnutrição grave (grau III) e sinais de desidratação (irritado, olhos encovados, fontanelas deprimidas, choro fraco e sem lágrimas, pulsos finos e elasticidade da pele diminuída), com descamação cutânea e hipotonia generalizadas, sem desconforto respiratório, anictérico, acianótico e afebril. Peso de 2.165g (percentil menor que 3). Exame físico segmentar sem anormalidades. O RN foi considerado desnutrido grau III e desidratado e internado na Unidade de Terapia Intensiva Neonatal do referido hospital sendo instituída hidratação venosa (HV) (80ml/ Kg/ dia) com taxa de infusão de glicose (TIG) de 5,0 mg/kcal/min e dieta oral por sucção (37 ml/ Kg/ dia) de LHO ou NANI 3/3 hs. A HV foi suspensa no 5º dia de internação. Os exames complementares realizados durante a internação constam no apêndice 1. Tendo em vista persistência dos vômitos, foi iniciado o leite Pregestimil por gastróclise e Motilium com melhora relativa. Do 5º ao 34º dia de internação, evoluiu com estabilidade clínica e vômitos esporádicos e dificuldade de ganho de peso (Apêndice 2 - gráfico do peso). No 35º dia, apresentou hipotonia, hiporreatividade, palidez cutâneo-mucosa e síndrome diarreica (fezes líquidas, 7 a 8 episódios por dia), necessitando de hidratação venosa. Foi considerada sepse e iniciado esquema empírico de antibioticoterapia (oxacilina e cefotaxime) mantidos por 10 dias. Evoluiu, nas duas semanas seguintes, apresentando hipoxemia corrigida com oxigenioterapia (no hood com FiO2 média de 0,3) que, por vezes, provocava episódios de hiperóxia. Havia também acidose metabólica necessitando de infusão de bicarbonato de sódio. Tentativas de diminuição ou retirada da infusão de bicarbonato de sódio produziram piora da acidose e, somente no 42º dia, foi possível sua retirada.
O screening para EIM ("teste do pezinho alargado ou completo" - perfil tandem), colhido no 37º dia de vida, mostrou resultado negativo para aminoacidopatias, distúrbios do ciclo da uréia e galactosemia, além das doenças do teste de screening padrão. Foi observado aumento moderado de C3, uma acilcarnitina com excreção urinaria aumentada em ambas acidemias metilmalônica e propiônica. Esta alteração pode ser encontrada na AMM ou acidemia propiônica. Foi então sugerida realização de análise quantitativa dos ácidos orgânicos urinários.
Aos 48 dias, seguindo orientação de especialista em EIM, foi iniciado ciclo de metronidazol por 5 dias, benzoato de sódio, biotina, tiamina, vitamina B12 e L-carnitina. Neste período, apresentou abalos de extremidades interpretados como crise convulsiva e iniciado dose de ataque de fenobarbital (10mg/ Kg) e manutenção de 5 mg/ Kg por 12 dias. Ocorreu, também, evento de desidratação grave e bradicardia com repercussão hemodinâmica necessitando de reanimação com ventilação com pressão positiva, masssagem cardíaca externa, adrenalina (3 doses), duas etapas rápidas de SF 0,9% (50 e 20 ml/ Kg), furosemida (3 doses), e reposição de bicarbonato de sódio. Persistia a acidose metabólica e necessidade de administração de bicarbonato de sódio.
Aos 49 dias de internação, foi iniciado o leite 80056® por gastróclise e em bomba de infusão contínua (BIC) por 30 minutos. Após três dias de seu início, voltou a apresentar intolerância à dieta. Foi tentado aumentar a taxa calórica diariamente associando também o triglicerídeo de cadeia média (TCM). Aos 52 dias de internação, iniciou-se coleta de gasometria arterial diariamente com o objetivo de manter o bicarbonato acima de 18 mosm/l. Iniciou-se o segundo ciclo de metronidazol por 5 dias e programado repetição a cada 20 dias. Aos 70 dias de internação, por apresentar edema periférico e palpebral, iniciou-se controle de proteínas plasmáticas, repondo albumina quando necessário. Evoluiu com alternância de desidratação e anemia e melhora relativa. No 84º dia foi iniciado o leite XMTVI analog na dose de 1g/ Kg/ dia com aumento progressivo de 0,5g/ Kg/ dia associado a NAN Soy e TCM. Evoluiu, entretanto, com persistência de acidose metabólica. No 89º dia, pesando 2.460g, apresentou episódio de hipotermia e bradicardia evoluindo para o óbito.
4 - REVISÃO DA LITERATURA
Muitos erros inatos do metabolismo (EIM) surgem precocemente, e, freqüentemente no período neonatal. Os principais grupos de EIM com aguda e severa apresentação na infância incluem: defeitos do metabolismo de carboidratos (galactosemia clássica), defeitos no ciclo da uréia (deficiência da transcarbamilase ornitina), acidúria orgânica (acidúria propiônica), aminoacidopatias (hiperglicemia não cetótica), defeitos no metabolismo de ácidos graxos (deficiência da cadeia média acil-CoA desidrogenase)13[i].
Acidúrias ou Acidemias Orgânicas são doenças hereditárias autossômicas recessivas causadas por deficiência severa da atividade de uma enzima, usualmente do metabolismo dos aminoácidos, resultando no acúmulo tecidual de um ou mais ácidos carboxílicos14[ii] 15[iii] (figura 1). Se comparada a outros grupos de EIM, as acidemias orgânicas são consideradas as doenças metabólicas mais freqüentes em crianças severamente enfermas16[iv] 17[v] e dos mais freqüentes grupos de enfermidades hereditárias do metabolismo18[vi]. A prevalência é provavelmente subestimada, uma vez que uma proporção substancial de casos permanece não diagnosticada ou diagnosticada erroneamente19[vii]. O quadro 1 demonstra a freqüência das acidemias orgânicas na população geral.
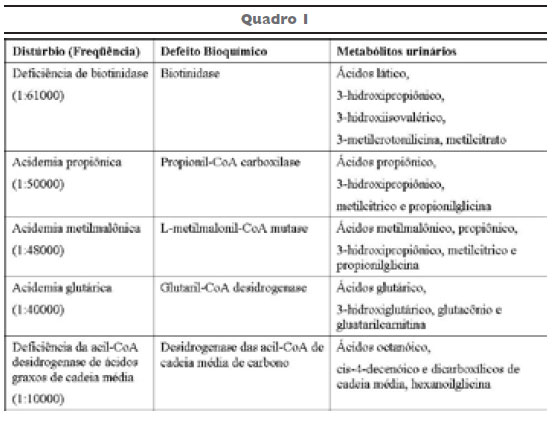
A acidemia metilmalônica (AMM) é um dos mais freqüentes erros inatos do metabolismo (EIM) dos ácidos orgânicos. O defeito bioquímico se localiza no metabolismo do propionato, na etapa de conversão de ácido metilmalônico a ácido succínico. Existem dois tipos de alterações básicas: o defeito na apoenzima (metilmalonil-mutase) e o defeito de coenzimas (metil B-12 e 5-desoxiadenosil-B12)1. A maioria dos pacientes tem um defeito na conversão de metilmalonil CoA a succinil CoA pelo defeito de apoenzima (metilmalonil-mutase). Esse grupo é o mais comum e não responde a administração de vitamina B12. No outro grupo, com defeito de coenzimas, a vitamina B12 (1mg/dia) reduz significativamente a excreção do ácido metilmalônico2 3 4 5 6 7.
A forma de apresentação mais comum da doença é a de início súbito no período neonatal. Cetoacidose, dificuldade de desenvolvimento e hipotonia muscular estão presentes nas duas formas da doença, seja com início no período neonatal ou mais tardiamente1 As convulsões mioclônicas podem levar ao coma e as crises de descompensação são precipitadas pela ingesta proteica, principalmente de leucina 2 3 4 ii[ix] iii[x].Os sintomas são geralmente graves e confundidos com septicemia devido a ocorrência de acidose e encefalopatia aguda em ambas as situações.8
De acordo com a figura 2, que ilustra um protocolo laboratorial para o diagnóstico das acidúrias orgânicas, o recomendado seria a análise dos ácidos orgânicos que é feito por cromatografia gasosa ou, de preferência, por cromatografia gasosa associada à espectrometria de massa20[viii] em amostras ocasionais de urina, devendo-se dar preferência à amostras colhidas durante crises de descompensação (Quadro 2). O diagnóstico correto de uma acidúria orgânica depende da identificação de vários metabólitos (ácidos orgânicos específicos). Muitas vezes, o diagnóstico é somente conseguido através da análise repetitiva de amostras coletadas em períodos distintos, especialmente durante descompensação metabólica, quando os níveis dos metabólitos anormais aumentam sua concentração. Quando o diagnóstico não é conseguido por esse método, podem ser realizados testes de sobrecarga com substratos proximais ao bloqueio metabólico ou, excepcionalmente, pela determinação da atividade enzimática em células cultivadas (fibroblastos)21[ix] 22[x].
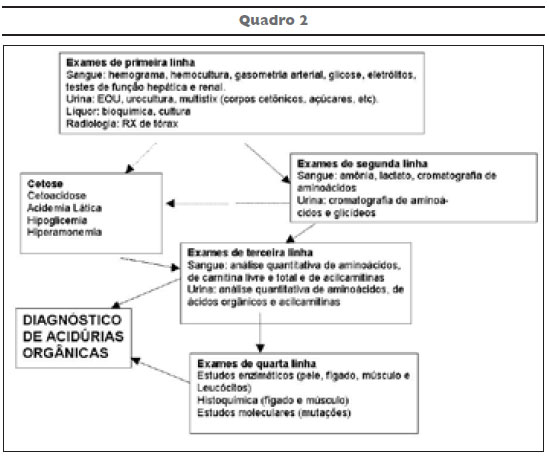
Em algumas ocasiões a criança afetada morre sem definição do diagnóstico da doença metabólica genética suspeita. Nestes casos, é essencial coletar amostras post mortem para esclarecimento diagnóstico e posterior aconselhamento genético e diagnóstico pré-natal. Os estudos mais importantes para essas situações são a determinação quantitativa de aminoácidos no plasma e líquido cefalorraquidiano, a determinação de ácidos orgânicos na urina e de acilcarnitinas em plasma, urina ou papel de filtro impregnado com sangue ou plasma8. O diagnóstico pré-natal em gravidezes de risco pode ser confirmado pela demonstração de diminuição significativa da atividade enzimática em vilosidades coriônicas e em fibroblastos23[xi].
O tratamento se baseia na restrição dos aminoácidos que se metabolizam a propionato e, em muitos casos, em preparados especiais que garantem o mínimo de proteína necessária para o crescimento1. As estratégias de tratamento estão resumidas no quadro 38.

O tratamento deve ser fundamentado na prevenção do acúmulo de substâncias tóxicas com a restrição de ingesta protéica ou de outros substratos e inibição do catabolismo com a prevenção de infecções, jejum prolongado ou abuso alimentar; na eliminação dos metabólitos tóxicos por exsanguíneo transfusão, hemodiálise e, por medidas de suporte geral (correção do pH sérico, ventilação mecânica assistida e hidratação adequada)24[xii].
Para o caso das doenças metabólicas com apresentação aguda neonatal, o tratamento deve iniciar o mais precocemente possível durante as crises metabólicas agudas, mesmo que a etiologia da doença seja ainda desconhecida. A terapia deve ser ampla e abranger vários grupos de doenças: acidúrias orgânicas, doenças do ciclo da uréia e defeitos no catabolismo de glicídeos e lipídeos. A retirada de toda a proteína da dieta durante as crises se faz necessária, uma vez que, a grande maioria das acidemias orgânicas é decorrente da falta de atividade enzimática em um passo do metabolismo dos aminoácidos. É também fundamental manter o anabolismo através da administração isocalórica ou hipercalórica (mais de 150 Kcal/Kg/dia) além da infusão intravenosa de grande quantidade de líquidos para evitar a desidratação. Sonda nasogástrica com alto aporte de alimentação lipídica é bastante útil. Com a melhora do paciente, deve-se adicionar gradualmente proteína até atingir uma quantidade tolerável que é individual para cada afetado. Megadoses de vitaminas específicas também são indicadas para os pacientes durante os episódios agudos, visto que o defeito metabólico em várias acidemias orgânicas pode ocorrer ao nível da formação das coenzimas, derivadas das vitaminas e essenciais para as reações enzimáticas25[xiii].
Alguns estudos têm recomendado o emprego de metronidazol como parte da terapêutica desta entidade26[xiv] 27[xv]. O uso desta droga e de outros agentes antibacterianos intestinas na acidemia metilmalônica é relativamente recente e apresenta resultados animadores26 27. A utilização desta droga se apóia na base teórica de tentar diminuir a produção de ácido metilmalônico pelas bactérias intestinais, síntese esta que alcança 30% do total28[xvi]. Espera-se com o tratamento a longo prazo, o aumento do apetite e ganho de peso além da diminuição da eliminação urinária de ácido metilmalônico26 27.
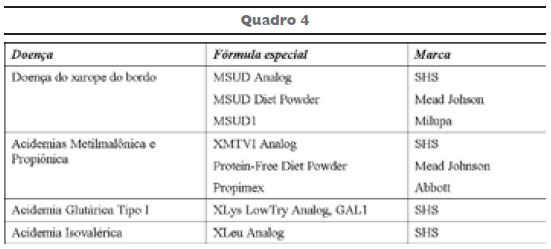
A terapia dietética de longo prazo é essencial para reduzir as concentrações dos metabólitos tóxicos e promover o desenvolvimento normal8. Suas metas visam estabilizar o balanço metabólico através da restrição dietética dos aminoácidos, isoleucina, metionina, treonina e valina, para manter a homeostase bioquímica. O aporte protéico inadequado pode resultar em déficit de crescimento em crianças, perda de peso, baixa concentração de albumina sérica, osteopenia em adultos e perda de cabelos em ambos. O aporte energético inadequado resulta em um déficit de crescimento e baixo peso. As fontes de energia recomendadas são: fórmula láctea de primeiro semestre, fórmula para acidemia para o primeiro ano (SHS XMTVI analog, Milupa OS1), polímeros de glicose, açúcar simples, saborizantes, triglicerídeos de cadeia média (TCM) e óleos vegetais.
O quadro 4 apresenta os produtos dietéticos comercialmente disponíveis para o tratamento específico de algumas dessas doenças8.
A determinação seriada da concentração urinária dos ácidos orgânicos é muito importante para verificar a adequação do tratamento dietético ou vitamínico. Através dos níveis plasmáticos de albumina e de aminoácidos, bem como a avaliação do desenvolvimento físico (peso e altura) pode-se monitorar o tratamento e o estado nutricional do afetado. A suplementação de L-carnitina (50 - 300mg/Kg/dia) tem sido bastante útil, pois os metabólitos tóxicos (ácidos) que se acumulam nas acidemias orgânicas se unem à carnitina para serem excretados, provocando déficit dessa substância que é essencial para o transporte de ácidos graxos para o interior da mitocôndria. A diminuição dos níveis teciduais de carnitina resulta, portanto, por um lado, na menor excreção desses metabólitos e, por outro, em déficit de produção de energia principalmente no músculo8.
Os recentes avanços no diagnóstico e tratamento dos EIM têm significativamente melhorado o prognóstico de muitas dessas doenças.
5 - DISCUSSÃO
Relatamos um lactente com quadro sugestivo de AMM, um EIM complexo, grave e isoladamente raro. É sabido que o diagnóstico das acidúrias orgânicas, na prática, não é realizado oportunamente e isto sempre leva a um pior prognóstico.
Neste caso, após o resultado do teste do pezinho alargado, excluiram-se as patologias relacionadas com o ciclo da uréia, aminoacidopatias e a galactosemia, tendo sido observado um aumento moderado de C3, que pode ser visto em ambas as acidúrias propiônica ou metilmalônica. De acordo com o protocolo diagnóstico, a análise quantitativa dos ácidos orgânicos urinários deveria ter sido realizada20. Porém, por dificuldades burocráticas, institucionais e governamentais, custo elevado do exame e indisponibilidade de laboratório, não foi possível realizá-la.
Deste modo, o tratamento foi iniciado para as acidúrias orgânicas, além da correção dos distúrbios hidroeletrolíticos e ácido-básicos com hidratação venosa, quando necessário, e reposição de bicarbonato de acordo com as gasometrias. Mesmo assim, o paciente evoluiu com piora progressiva, necessitando de reposição contínua de bicarbonato de sódio. Evoluiu também com episódios de edema periférico e periorbitário, sendo necessária a reposição de albumina. Os ciclos de metronidazol, a suplementação de L-carnitina e megadoses de vitaminas foram feitos, de acordo com o protocolo recomendado para tratamento de AMM26 27.
Apesar de todos os esforços, com a fórmula láctea iniciada tardiamente, persistiu a acidose metabólica e o paciente nem chegou a recuperar o peso de nascimento, evoluindo a óbito.
Ressaltamos as dificuldades do manuseio do paciente com um EIM complexo como a AMM. A maior dificuldade do caso foi obter o aporte nutricional adequado para o paciente. A fórmula láctea recomendada, XMTVI analog8, foi iniciada tardiamente. Diante da demora, foi tentada, sem sucesso, a associação do leite 80056 e TCM.
Acreditamos que essas dificuldades devem ter contribuído para o desfecho. Apesar de ter sido seguido o protocolo de tratamento8, a impossibilidade de cumprir o protocolo de acompanhamento8 associada à não disponibilidade do leite recomendado foram os fatores mais influentes.
6 - CONSIDERAÇÕES FINAIS
Conhecer a AMM é importante, pois o prognóstico do paciente está relacionado ao tempo gasto para se chegar ao diagnóstico e, assim, instituir o tratamento adequado. As dificuldades de se seguir o protocolo diagnóstico e terapêutico contribuem para um pior desfecho. É essencial que o pediatra se familiarize com esta desordem, para que este diagnóstico seja aventado diante de situações clínicas semelhantes.
REFERÊNCIAS BIBLIOGRÁFICAS
1. [i] SANJURIO CRESPO P, LABAYRU ECHEVERRIA M, INGUNZA AGUIRRE N, SASIETA ALTUNA M: Uso de metronidazol em cuatro casos de acidemia metil-malónica. An. Esp. Pediatr, 38(5),9449-451,1993.
2. [ii] MELENGELLO J: Pediatria. 5 Ed.: Editora Panamericana. Buenos Aires, 1999.
3. [iii] VOLPE J. Neurology of the newborn: 4 ed. Philadelphia: WB Saunders; 2000.
4. [iv] MENKES J. Child Neurology: 2 ed. Philadelphia: Lea & Febiger; 1980.
5. [v] MORENO-FUENMAYOR H, CHACIN C, LAGUNA E, GONZÁLEZ B: Acidosis metabólica com hiperglicinemia e hiperglicinuria y repuesta clínica a la vitamina B12. Arch Venez Puer Ped 45:130-7,1982.
6. [vi] CHENEL C, WOOD C, GOURRIER E, ZITTOUN J, CAJADEWALL L, OGIER H: Neonatal hemolytic-uremi syndrome, methylmalonic acidúria and homocystinuria caused by intracellular vitamin B12 deficiency. Value of etiological diagnosis. Arch Fr Pediatr 50:749-54,1993.
7. [vii] GRUSZCZYNSKA B, MIELNICZUK Z, KATYNSKA B, PRONCEKA E: A case of vitamin B12-independent methylmalonic acidúria. Mater Med Pol 23:219-22,1991.
8. [viii] CARMEN RV, WAJNER M: Acidúrias orgânicas: diagnóstico e tratamento. Artigo de Revisão. Revista AMRIGS, Porto Alegre, 45(1,2):77-82,2001.
9. [ix] CAMPISTOL J: Errores congénitos del metabolismo intermediario con repercusión neurológica. Aminoacidopatias. Acidúrias Orgânicas. In Aparício JM, Artiga J, Campistol J, Campos J, Casas C, Castro-Gago M, et al, eds. Neurologia pediátrica. Barcelona: Égon; p. 95-113,2000..
10. [x] SWAIMAN KF. Neurologia pediátrica. 2ed. St. Louis: Mosby/Doyma; 1996.
11. [xi] Massachusetts Department of Public Health: Newborn screening for metabolic disorders. N Engl J Med 288:1299-1300,1973.
12. [xii] BURTON BK: Inborn errors of metabolism in infancy: a guide to diagnosis. Pediatrics 102:e69,1998.
13. [xiii] DESNICK RJ: Treatment of Genetic Disease. New York, Churchill Livingstone Inc., 1991.
14. [xiv] THOMPSON, G N; CHALMERS, R A; WALTER, J H; BRESSON, J L; LYONNET, S L; REED, P J; SAUDUBRAY, J M; LEONARD, J V, y HALLIDAY, T: The use of metronidazole in management of methylmalonic and propionic acidemias. Eur J Pediatr; 149:792-796,1990.
15. [xv] KOLETZKO, B; BACHMANN, C, y WENDEL, U: Antibiotic therapy for improvement of metabolic control in methylmalonic acidúria. J Pediatr; 117:99-101,1990.
16. [xvi] BAIN, M; JONES, M; BORRIELO, S P; REED, P J; TRACEY, B M; CHALMERS, R A, y STACEY, T E: Contribution of gut bacterial metabolism to human metabolic disease. Lancet 1:1078-1079,1998.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()