Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 10(1) - Maio 2009
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
- Outros do Autor
Revisoes em Pediatria
Doença de Von Gierke: estudo de revisão
Von Gierke disease: revision study
Frederico Henrique Salles Nunes
Graduaçao (Nutricionista). Fundaçao Hospitalar do Estado de Minas Gerais - FHEMIG - Hospital Infantil Joao Paulo II.
Endereço para correspondência:
Frederico Henrique Salles Nunes
Rua Nilo 121A, Bairro: São Salvador
Belo Horizonte, MG
e-mail: frederico-nunes@ig.com.br
Resumo
As glicogenoses são doenças de armazenamento de glicogênio e a doença de von Gierke é a mais comum delas, sendo que as complicações metabólicas da doença incluem hipoglicemia, acidemia láctica, hiperuricemia, hipofosfatemia e adenoma hepático. Nesse trabalho foi feita uma revisão dos conhecimentos atuais acerca da doença Von Gierke com suas características e tratamento aplicado. Verificou-se que a adesão ao tratamento é de vital importância para o controle da doença.Palavras-chave: Doença de von Gierke, Glicogenoses, Nutriçao.
Abstract
The glycogenosis are glycogen storage diseases, the von Gierke disease is the more common of them, the metabolic complications of the disease include, hypoglycemia, lactic acidemia, hyperuricemia, hypercholesterolemia and hepatic adenoma. Our goal with this study was to make a review of the current knowledge concerning the disease von Gierke your characteristics and applied treatment. The adhesion to the treatment is of vital importance for the control of the disease.Keywords: Von Gierke disease, Glycogenosis, Nutrition.
INTRODUÇÃO
Como todos os erros inatos de metabolismo são individualmente raros e têm apresentações clínicas inespecíficas é comum o pediatra cogitá-los tardiamente1. Diagnosticar rapidamente é essencial para impedir o agravamento e a irreversibilidade dos sintomas, podendo representar a vida do paciente em alguns casos2.
OBJETIVO
Foi objetivo do trabalho atual, fazer uma revisão sobre a glicogenose tipo Ia, doença de depósito de glicogênio resultante da deficiência da enzima glicose-6-fosfatase com o intuito de dotar os pediatras, nutricionistas e demais profissionais da saúde de informações necessárias para o diagnóstico e tratamento adequado dos indivíduos acometidos por essa doença.
METODOLOGIA
A revisão de literatura foi realizada através da seleção de artigos da base de dados Medline, Scielo e Google Acadêmico, com os artigos de periódicos médicos e nutricionais nacionais e internacionais mais importantes dos últimos vinte anos e livros nutricionais e médicos com informações relevantes sobre a glicogenose tipo Ia.
REVISÃO
DEFINIÇÃO
As Doenças Metabólicas Hereditárias são causadas por erros inatos do metabolismo e resultam da falta de atividade de uma ou mais enzimas específicas ou defeitos no transporte de proteínas3.
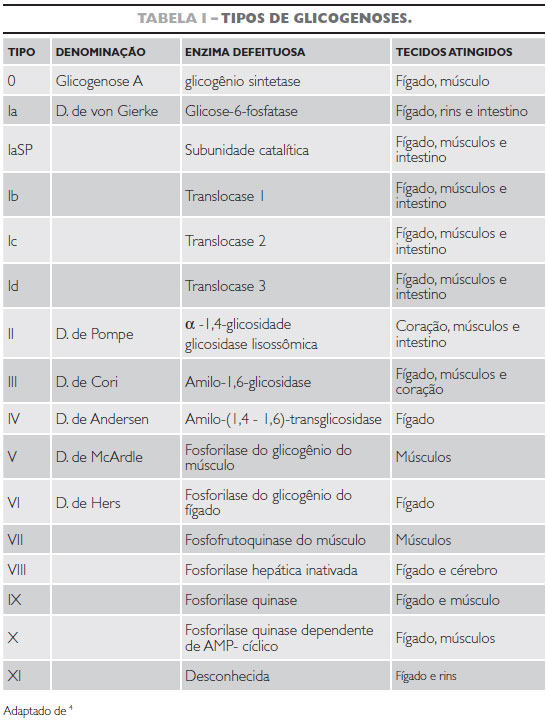
São denominadas glicogenoses as afecções decorrentes de erro metabólico hereditário que resulta em anormalidade da concentração e/ou da estrutura do glicogênio em qualquer tecido do organismo4.
A glicogenose tipo I é caracterizada pela deficiência de glicose-6-fosfatase (G-6-Pase), uma enzima composta por um sistema multicomponente que compreende o sítio ativo na superfície luminal do retículo endoplasmático e três translocases4. A glicogenose tipo Ia envolve a deficiência da unidade catalítica que é responsável por desfosforilar a glicose-6-fosfato1,5.
A entidade foi denominada doença de von Gierke em homenagem ao seu descobridor Edgar von Gierke4. Os tipo de glicogenose são descritos na tabela I.
DIAGNÓSTICO
A clínica que alia a história da doença e um bom exame físico contribui imensamente para um diagnóstico preciso através da identificação de sinais característicos de cada grupo de erros inatos do metabolismo2,6.
Os primeiros sinais e sintomas que podem levar a investigação da presença de glicogenoses em recém-nascido são: quadro neurológico grave com manifestação precoce de convulsão e aumento de ácido láctico no sangue, hipoglicemia, convulsão, acidose metabólica, hiperuricemia e hepatomegalia4,7.
A doença é descrita nos primeiros 28 dias de vida (período neonatal) quando os bebês costumam apresentar hipoglicemia após pequenos períodos de jejum ou após infecções. A hipoglicemia se caracteriza por palidez, suor frio e convulsões. Ao exame físico nota-se o aumento hepático, a obesidade troncular e a "face de boneca"7.
O diagnóstico pela determinação da atividade enzimática só pode ser realizado em órgão com grande expressão da enzima exigindo uma biópsia hepática.
São descritas atualmente 14 alterações genéticas diferentes capazes de alterar ou inativar a atividade da G-6-Pase causando a doença de von Gierke, 5,9 podendo o diagnóstico baseado no DNA ser realizado em qualquer tecido disponível como sangue, pele ou em caso de teste pré-natal vilosidade corionica e aminiócitos10-12.
A glicose-6-fosfatase é codificada pelo gene 17, LEI em estudo descreve que a informação para a codificação da enzima ocupa um espaço de 12,5 Kb e consiste de cinco exons, desse modo podem ocorrer varias alterações capazes de afetar a atividade da enzima7,13-15.
EPIDEMIOLOGIA
A glicogenose tipo Ia é de herança genética autossômica recessiva, ou seja, têm um risco de ocorrência de 25% a cada gestação de pais heterozigotos, apresentando incidência de 1:100.000 nascidos vivos3,16. Essa incidência pode estar subestimada, pois estudos indicam que muitas crianças morrem antes do diagnóstico da doença.
A glicogenose tipo Ia representa cerca de 25% das glicogenoses4.
FISIOPATOLOGIA
O glicogênio é um polímero de armazenamento encontrado no citosol de todas as células do organismo, entretanto está em maior concentração no fígado e músculo esquelético,17 a formação deste permite o acúmulo de glicose nas células sem aumentar a pressão osmótica dentro destas18.
A degradação do glicogênio envolve três etapas: a liberação de glicose 1-fosfato do glicogênio; remodelação do glicogênio para que seja possível uma nova degradação e a conversão da glicose-1-fosfato em glicose-6-fosfato, para sua devida metabolização17.
O glicogênio é um polímero de glicose ramificado unido através de ligações glicosidicas α-1,4 em sua cadeia linear e α-1,6 em suas ramificações que ocorrem a cada oito a dez unidades de glicose em cadeia linear, por isso a degradação do glicogênio necessita de varias enzimas. A fosforólise das ligações α-1,4 gera glicose-1-fosfato, já a quebra das ligação α-1,6 libera uma glicose livre, esse mecanismo produz cerca de 90% de glicose-1-fosfato e 10% de glicose livre19,20.
A glicose-6-fosfato então pode seguir três destinos: ser usada como substrato energético na glicólise, seguir pela via das pentoses-fosfato e gerar NADPH e derivados de glicose ou ser convertida à glicose livre sendo liberada na corrente sanguínea17. A glicose fosforilada (glicose-6-fosfato) não pode atravessar a membrana e difundir-se para fora da célula hepática, para a liberação da glicose é preciso a ação da enzima glicose 6-fosfatase que cliva a fosforila formando glicose livre e ortofosfato17.
O metabolismo de glicogênio e os possíveis destinos da glicose são mostrados na figura I.
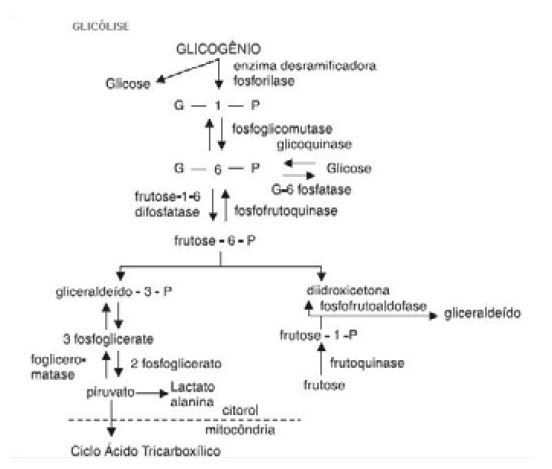
Figura 1 - Metabolismo de glicogênio. - Adapatado de 4
A glicose 6-fosfatase está localizada no lado da luz da membrana do reticulo endoplasmático liso para onde a glicose 6-fosfato é transportada e então hidrolisada à ortofosfato e glicose livre pela unidade catalítica da glicose-6-fosfatase19.
A doença de von Gierke se apresenta quando a atividade da unidade catalítica da glicose 6-fosfatase está ausente ou com atividade reduzida no fígado, rim e intestino.
QUADRO CLÍNICO E LABORATORIAL
HIPOGLICEMIA
Com a deficiência da glicose-6-fosfatase a glicose-6-fosfato principal produto da glicogenólise e gliconeogenese não pode ser convertida em glicose livre, o que torna impossível a travessia desta pela membrana citoplasmática. Como consequência apenas 8 a 10% da glicose retirada do glicogênio pode ser usada para o controle da glicemia, sendo que essa glicose é proveniente da quebra das ligações α-1,617. Com isso o fígado não é capaz de manter os níveis de glicose no sangue normais, ocorrendo a hipoglicemia.
Há relatos que com o aumento idade há uma tendência para a diminuição da hipoglicemia pela redução do consumo energético4.
ACIDOSE LÁCTICA
A presença de glicose-6-fosfato em excesso no fígado dispara a glicólise, com o excesso de piruvato produzido o tecido não pode ser suprido de oxigênio suficiente para suportar toda a oxidação aeróbica desse nutriente. Com isso utiliza-se o metabolismo anaeróbico com produção de lactato, levando ao aumento dos níveis de lactato e piruvato no sangue17.
Em situações normais o lactato é convertido no fígado em glicose por um ciclo de reações chamado de ciclo de Cori, realizando o processo de gliconeogênese, porém isso não ocorre na glicogenose tipo Ia, aumentando os níveis de lactato cerca de quatro vezes acima dos valores normais21.
Mesmo com o tratamento ideal em que a glicose é mantida a níveis normais, a concentração de lactato não diminui a níveis de 3 a 5 mmoles/L valores considerados normais.
HIPERURICEMIA
A hiperuricemia resulta tanto da diminuição da depuração renal de urato secundária à competição com o ácido láctico e outros, quanto do aumento da produção do ácido úrico4,22.
A glicose-6-fosfato acumulada é desviada para a via das pentoses fosfato, levando ao aumento da síntese de purinas, com a grande produção os produtos purínicos são degradados até ácido úrico21.
A degradação do ATP se acelera em resposta à hipoglicemia e ao aumento dos níveis de glucagon e sua ressíntese requer glicose. Com a baixa disponibilidade de glicose livre para a distribuição aos tecidos, ocorre acúmulo de ADP que é convertido a ácido úrico4.
A gota, os cálculos renais e a nefropatia são as consequências da hiperuricemia, a doença renal crônica de etiologia desconhecida também é uma complicação comum da glicogenose tipo Ia incluindo fibrose intersticial, atrofia tubular, focal e segmentar e glomeruloesclerose4,23.
HIPOFOSFATEMIA
Uma das consequências metabólicas da deficiência da G-6-Pase é a hipofosfatemia observada durante os episódios hipoglicêmicos, pois devido à impossibilidade de conversão da glicose-6-fosfato em glicose, o fosfato não é liberado da molécula de glicose-6-fosfato, resultando em baixa de fosfato intracelular livre. Como há necessidade do fosfato para outras atividades, ocorre desvio compensatório do fósforo extracelular para o interior da célula gerando hipofosfatemia4,24,25.
HIPERLIPIDEMIA
Os níveis de triglicérides podem chegar a 4.000 a 6.000 mg/dl e o colesterol pode atingir 400 a 600 mg/dl com LDL elevado e HDL reduzido.
A hiperlipidemia se deve ao aumento dos produtos da via glicolítica como o NADPH, NADH, fosfato, glicerol-3-fosfato e coenzima A, essenciais para a síntese de colesterol e ácidos graxos. Os triacilgliceróis são sintetizados a partir de um ácido graxo ativado e um produto fosforilado de três carbonos, proveniente do catabolismo da glicose4,19.
A hepatomegalia acentuada característica da glicogenose é atribuída mais à deposição de gordura no fígado do que ao acúmulo de glicogênio, causando tumefação dos hepatócitos4. Porém mesmo com o perfil lipídico alterado os pacientes mostram risco de doença coronariana e isquêmicas iguais a de um individuo normal4.
O manejo da dieta associa-se com a queda dos níveis de triglicérides, mas estes não chegam a alcançar valores normais e a associação de clofibrato e niacina obtém sucesso no controle da trigliceridemia em pacientes que não responderam à dieta.
COMPLICAÇÕES RENAIS
Pacientes com glicogenose tipo Ia sob bom controle metabólico da glicemia ainda experimentam a longo prazo complicações da doença, eles continuam a sofrer de hiperlipidemia, hiperuricemia, hipercalciúria e acidemia láctica e doença renal crônica de etiologia desconhecida23.
A nefropatia por gota e a nefrocalcinose podem estar presentes. A hipercalciúria provavelmente é um fenômeno secundário a alterações da acidificação renal4,26.
Pacientes com glicogenose tipo I também manifestam hiperuricemia e hiperlipidemia que podem ser fatores independentes de risco para o desenvolvimento e a progressão da doença renal23.
A disfunção renal é vista na maioria dos pacientes a partir da segunda década de vida4.
ADENOMAS E NEOPLASIAS HEPÁTICAS
A partir da segunda e terceira década de vida cerca de 50 a 70% dos indivíduos com glicogenose tipo I desenvolvem adenomas hepáticos, que podem levar a hemorragia aguda e transformação maligna, os tumores são múltiplos, pequenos e não capsulados4.
Os pacientes do sexo masculino apresentam adenomas hepáticos na proporção de 2:1 em relação ao sexo feminino, sendo que a patogênese dos carcinomas em geral não esta elucidada, porém existem algumas hipóteses que incluem desequilíbrio da relação glucagon/insulina, sobrecarga de glicogênio celular e ativação de protooncogenes4.
TRATAMENTO
O tratamento da doença de von Gierke é feito principalmente através da dieta. Os objetivos da dietoterapia são manter a homeostase da glicose para prevenir as reações hipoglicêmicas e fornecer proteínas e calorias suficientes para o balanço positivo de nitrogênio e para o crescimento normal evitando déficit de crescimento e as demais alterações metabólicas27.
Deve-se atingir a Recommended Dietary Allowances (RDA). A distribuição de macronutrientes da dieta deve ter percentual de 30 a 45% de carboidrato oferecido na forma de amido de milho cru e os carboidratos totais deve atingir de 60 a 70% do total energético da dieta. A distribuição de lipídeos deve abranger de 20 a 25% das calorias sendo seguida a mesma proporção de uma dieta comum para as gorduras monoinsaturada, poliinsaturada, saturada e colesterol, a proteína deve atingir de 10 a 15% 4,16,27.
A frutose e a galactose são metabolizados à glicose-6-fosfato, portanto deve-se evitar o consumo de lactose, frutose e sacarose, que podem provocar acidose láctica, no entanto não há concordância cientifica sobre a restrição desses monossacarídeos4,16.
Há duas abordagens dietoterápicas tradicionais para manter os níveis de glicemia dentro dos valores normais de referência; uma delas é alimentação contínua por sonda que é considerada o maior avanço no tratamento da glicogenose, a administração oral de polímeros de glicose de digestão lenta é outra conduta adotada para a manutenção dos níveis de glicemia podendo-se ainda fazer a combinação das duas técnicas4,16. Tanto através do tratamento com amido de milho cru como a Infusão noturna de glicose (ING) consegue-se bons resultados no controle da glicemia e da taxa de crescimento4.
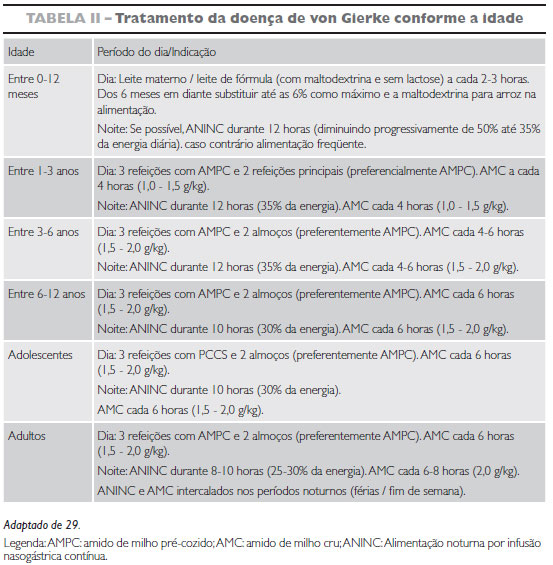
O uso do amido de milho cru para a manutenção da glicemia é recomendado a cada 2 a 4 horas, dependendo do intervalo de tolerância ao jejum relativo à atividade residual da enzima, em menores de 2 anos deve-se diluir 1,65 g/kg de peso/dose de amido de milho em 1:2 partes de água à temperatura ambiente, e para crianças maiores de 2 anos em intervalos de quatro horas na dose de 1,75 a 2,5g por kg de peso por dose. O amido deve ser dado com água fria, pois a mistura com água morna, quente ou limonada acelera sua hidrólise, não obtendo o efeito desejado4,8,28.
O início da ING é realizado no hospital com taxa de infusão de glicose igual a 7mg/kg/min e posteriormente ajustada para 4-6mg/kg/min para manter a glicemia recomendando a solução de dextrose a 25% ou 50 %. Na primeira noite a glicemia precisa ser controlada a cada quatro horas e antes da retirada a cada manhã. Para a infusão noturna de glicose é necessária a realização do cálculo da taxa basal de produção de glicose: y = 0,0014x3 - 0,214x2 + 10,411x - 9,084, onde y = mg de glicose por minuto e x = peso em kg, o resultado representa a quantidade mínima de glicose necessária para manter a glicemia em níveis normais 4,16.
A transição entre a ING e a alimentação e vice-versa deve ser bem controlada para impedir a hipoglicemia, a última refeição deve ser oferecida não ultrapassando três horas para o inicio da ING, já a primeira refeição deve ser oferecida antes de se interromper a ING, com antecedência mínima de 30 minutos antes da interrupção4,16.
Entre 4 e 6 meses de idade é indicada a introdução de outros alimentos como é habitual na idade, porém carboidratos complexos devem ter ênfase na forma de aveia, cevada, arroz, massas e legumes, preterindo batatas e pães 4,16.
O uso do amido de milho cru é recomendado após os dois anos de idade, pois das enzimas amilase pancreática e a glicoamilase intestinal responsáveis pela digestão de amido no organismo só a glicoamilase apresenta atividade comparada a de um adulto a partir do primeiro mês de vida, a atividade da amilase pancreática no período neonatal é considerada desprezível, só alcançando níveis comparados ao de um adulto aos dois anos de idade. No entanto sua atividade pode ser estimulada a partir do oitavo mês de vida, antes disso as crianças podem não apresentar digestão adequada do amido4,16.
O plano dietético deve ser feito cuidadosamente e revisado para proporcionar aqueles nutrientes essenciais, merecendo atenção especial o cálcio, devido ao limitado consumo de leite e a vitamina D. Além disso, o aumento do metabolismo dos carboidratos necessita de grandes quantidades de vitamina B129.
A dietoterapia apresenta-se como um eficiente método para controle da doença. A avaliação do sucesso do tratamento pode ser realizada através dos níveis de lactato e acido úrico no sangue, o nível sérico de lactato reflete o estado metabólico recente decorrente da queda da glicemia no dia, enquanto que os níveis do ácido úrico e dos triglicérides refletem o controle a longo prazo16.
O tratamento para portadores da doença de von Gierke conforme a idade é apresentado na TABELA II.
O tratamento dietético da glicogenose é acompanhado da melhora das alterações renais. A rápida resposta ao tratamento pode explicar a razão pela qual a disfunção renal não é encontrada mais frequentemente4.
O tratamento medicamentoso pode ser usado nos casos em que a dietoterapia não é capaz de evitar as complicações metabólicas. O alopurinol pode ser usado em casos em que não se consegue controlar os níveis de ácido úrico8.
Em casos de hipoglicemia grave deve-se administrar imediatamente uma solução de glicose por via intravenosa29.
O transplante hepático pode ser considerado nos casos em que o manejo dietético não obtém sucesso, necessitando de frequentes internações. O transplante de fígado é considerado o procedimento mais complexo da cirurgia moderna, pois interfere em muitas funções do organismo. O sucesso da operação depende de uma boa infra-estrutura hospitalar, uma experiente e bem treinada equipe multiprofissional. 27,30.
O transplante hepático é utilizado com sucesso nos casos de adenoma hepático com aumento gradativo, quando não se pode afastar a possibilidade da transformação maligna e que a ressecção esteja inviabilizada4.
PROGNÓSTICO
O descumprimento do tratamento resulta em sérias consequências metabólicas, com graves crises de hipoglicemia acompanhada de danos neurológicos4,31.
O tratamento proposto atualmente apresenta significante melhora do quadro clínico, sendo refletido na melhora do prognóstico nos pacientes com glicogenose tipo Ia, com expectativa de vida ultrapassando a terceira década4.
Devido à relativa raridade da doença não existem dados sobre a porcentagem de sobrevida de acordo com as diferentes faixas etárias.
CONCLUSÃO
O diagnóstico precoce dos erros inatos do metabolismo é de extrema importância para o inicio imediato do tratamento, sendo necessário conhecer a fisiopatologia para a adoção da dietoterapia ideal e para o indispensável aconselhamento familiar que compreenda principalmente, o prognóstico do paciente e a importância da adesão ao tratamento.
AGRADECIMENTO
Agradeço a Dr. Neuza Maria de Magalhães pelas opiniões criticas e incentivo na realização do trabalho e a Nutricionista do Hospital Infantil João Paulo II, Olindina Neme Barbosa Miranda pelas informações disponibilizadas.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Jardim LB, Ashton-Prolla P. Erros inatos do metabolismo em crianças e recém-nascidos agudamente enfermos: guia para o seu diagnóstico e manejo. J Ped 1996;72:63-72.
2. El Husny AS, Fernandes-Caldato MC. Erros inatos do metabolismo: revisão de literatura. Rev Para Med 2006;20(2):41-5.
3. Martins AM, Micheletti C, D'Almeida V, Frangipani BJ. Erros inatos do metabolismo: abordagem clinica. [período na internet] 1998 [citado 2008 Out 07]. Disponível em: http://www.lagem-ba.com/publicacoes/artigos/eim/eim01.pdf.
4. Reis CVS, Penna FJ, Oliveira MCC, Roquete MLV. Glicogenose tipo I. J Ped 1999;75:277-35.
5. Chou JY, Mansfield BC. Mutations in the Glucose-6-Phosphatase- ± (G6PC) Gene that Cause Type Ia Glycogen Storage Disease. Hum Mut 2008;29:921-30.
± (G6PC) Gene that Cause Type Ia Glycogen Storage Disease. Hum Mut 2008;29:921-30.
6. Feferbaum R, Ceccon MEJ, Diniz EMA, Leone CR, Costa MTZ, Corradini HB. Erros inatos de metabolismo: Uma abordagem diagnóstica. Ped 1994;16:82-6.
7. Brunoni D. Avaliação Genético-Clínica do Recém-Nascido. [período na internet] 2007 Set [citado 2008 Out 07]. Disponível em: http://www.sbgm.org.br/cfm/Av_Gen_Clin_Recem_Nascido.pdf.
8. Tommaso AM. A de. Glicogenoses. [período na internet] 2000 [citado 2008 Out 09]. Disponível em: http://http://www.hepcentro.com.br/glicogenoses.htm.
9. Stroppiano M, Regis S, Dirocco M, Caroli F, Gandullia P, Gatti R. Mutations in the glucose-6-phosphatase gene of 53 Italian patients with glycogen storage disease type Ia. J Inher. Metab. Dis 1999;22:43-9.
10. Cleary MA, Wraith JE. Antenatal diagnosis of inborn errors of metabolism. Arch Dis Child 1991;66:816-22.
11. Winchester B. Prenatal diagnosis of enzyme defects. Arch Dis Child 1990;65:59-67.
12. Lei KJ, Chen YT, Chen H, Wong LC, Liu J. Genetic Basis of Glycogen Storage Disease Type 1a: Prevalent Mutations at the Glucose-6-Phosphatase Locus. Am J Hum. Genet 1995;57:766-71.
13. Chevalier-Porst F, Bozon D, Bonardot A, Bruni N, Mithieux G, Mathieu M. Mutation analysis in 24 French patients with glycogen storage disease type 1a. F Med Genet 1996;33:358-60.
14. Lei KJ, Pan C, Shelly LL, Liu J, Chou JY. Identification of mutations in the gene for glucose-6-phosphatase, the enzyme deficient in glycogen storage disease type 1a. J Clin Invest 1994;93:1994-9.
15. Departamento de Nutrição da Sociedade Brasileira de Pediatria. Temas de nutrição em pediatria. Rio de Janeiro, 2004.
16. Basso, L. S.; Speridião, P. da G. L.; Fagundes Neto, U. Terapia Nutricional nas glicogenoses. [período na internet] 2006 [citado 2008 Out 07]. Disponível em: http://www.e-gastroped.com.br/sep06/glicogenoses.htm.
17. Berg JM, Tymoczko JL, Stryer L. Bioquímica. 4. ed. Rio de Janeiro, Guanabara Koogan, 2004.
18. Fontes R. Metabolismo do Glicogênio. [período na internet] 2007 [citado 2008 Out 07]. Disponível em: http://users.med.up.pt/ruifonte/PDFs/2006-2007/G08_MetGlicogenio.pdf.
19. Devlin TM. Manual de bioquímica com correlações clínicas. 4. ed. São Paulo, Edgard Blucher, 1998.
20. Fontes R. Gliconeogénese e Metabolismo de Glicogênio. [período na internet] 2002 [citado 2008 Out 07]. Disponível em: http://2002-2003/Nut/BII/TXT-Gliconeogenese_MetGlicogenio.pdf.
21. Nelson DL, Cox MM. Lehninger Princípios de bioquímica. 3. ed. São Paulo, Sarvier, 2002.
22. Leal V O, Leite Júnior M, Mafra D. Acidose metabólica na doença renal crônica: abordagem nutricional. Rev Nutr 2008;21:93-103.
23. Yiu WH, Chen YT, Chen H, Wong LC, Liu J. The angiotensin system mediates renal fibrosis in glycogen storage disease type Ia nephropathy. Kidney Int 2008;73:716-23.
24. Araujo IM, Guilhen JC, Marques JA, Pavanetti LC. Correção da hipofosfatemia grave com dialisato enriquecido de fosforo em paciente hemodialisado. Arq Ciênc Saúde 2006;13:220-2.
25. Lauar JT, Araújo LHL, Fialho ÉL, Gazolla MVB, Miguel RCC. Associação entre hipofosfatemia e alcoolismo. J Bras de gastroenterol 2006;6:38-40.
26. Bastos, M. Doença renal crônica: Problemas e soluções. [período na internet] 2004 [citado 2008 Out 07]. Disponível em: http://www.sbn.org.br/JBN/26-4/v26e4p202.pdf.
27. Bodinski LH, Ritt R. Dietoterapia nos erros inatos do metabolismo. In: Bodinski LH, Ritt R. Dietoterapia Princípios e pratica. São Paulo: Atheneu, 2006. p. 282-3.
28. Martins AM, Micheletti C, Vertemati T. Erros inatos de metabolismo grupo 3. [período na internet] 2007 [citado 2008 Nov 23]. Disponível em: www.unirio.br/dmp/Graduacao/Medicina/Patologia/Erros%20Inatos%20do%20Metabolismo.pdf.
29. Rake JP. Estudio Europeo sobre Glucogenosis Tipo I. Rijksunniversiteit [período na internet] 2006 Set [citado 2008 Out 07]. Disponível em: www.glucogenosis.org/docs/ESGSDI.pdf.
30. Mies S. Transplante de fígado. Revista Associação Médica Brasileira, 1998;44:127-34.
31. Rennie J, Boylan G. Tratamento das convulsões neonatais. 2007. Arc Dis Child Fetal Neonatal 2007;92:148-50.
AVALIAÇÃO
Questões artigo "Doença de von Gierke: estudo de revisão".
1. Dentre os sinais e sintomas que podem levar a investigação da presença de glicogenoses em recém-nascido são, exceto:
a) Convulsão
b) Hepatomegalia
c) Hiperglicemia
d) Face de Boneca
2 A doença de von Gierke pode ser confirmada pelos seguintes
exames, exceto:
a) Determinação da atividade enzimática de glicose-6-fosfatase em tecido hepático
b) Ausência de glicose-6-fosfatase no sangue
c) Analise de DNA, detectando-se uma das alterações do gene 17, compatível com as alterações encontradas na doença de von Gierke
d) Teste pré-natal de vilosidade corionica e aminiócitos
3. Em relação à dietoterapia para a doença de von Gierke é correto afirmar que:
a) A frutose e a galactose são metabolizados à glicose-6-fosfato, portanto deve-se evitar o consumo de lactose, frutose e sacarose, que podem provocar hipofosfatemia
b) A infusão noturna de glicose é o tratamento ideal, indicado a todos os paciente
c) Não se deve atingir a Recommended Dietary Allowances (RDA) dos nutrientes.
d) O amido deve ser dado com água fria, a mistura com água morna, quente ou limonada acelera sua hidrólise, não obtendo o efeito desejado.
Preencher ficha na página 43 e enviar à SOPERJ

![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()