Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 24(4) - Dezembro 2024
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Artigo Original
Cardiopatias congênitas com repercussão hemodinâmica pós-natal diagnosticadas pelo ecocardiograma fetal - Parte 1
Congenital heart diseases with postnatal repercussion diagnoses by fetal echocardiogram - Part 1
Anna Esther Araujo e Silva1,2; Eliane Lucas1,3; Maria de Fátima Monteiro Pereira Leite1,4; Nathalie Jeanne Bravo-Valenzuela1,5; Talita Nolasco Loureiro1,6
DOI:10.31365/issn.2595-1769.v24i4p109-118
1. SOPERJ, Departamento de Cardiopediatria - Rio de Janeiro - RJ - Brasil
2. Hospital Universitário Antônio Pedro, Universidade Federal Fluminense, Departamento de Pediatria - Niterói - RJ - Brasil
3. Faculdade de Medicina de Teresópolis (UNIFESO), Departamento de Pediatria - Teresópolis - RJ - Brasil
4. Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira - IFF- FIOCRUZ, Departamento de Pediatria - Rio de Janeiro - RJ - Brasil
5. IPPMG, Universidade Federal do Rio de Janeiro, Departamento de Pediatria - Rio de Janeiro - RJ - Brasil
6. Hospital Universitário Pedro Ernesto, Universidade do Estado do Rio de Janeiro, Departamento de Pediatria - Rio de Janeiro - RJ - Brasil
Endereço para correspondência:
Correspondência:mfatima2@yahoo.com
Instituição: SOPERJ, Departamento de Cardiopediatria - Rio de Janeiro - RJ - Brasil
Revisado por: Em breve
Recebido em: 27/08/2024
Aprovado em: 24/09/2024
Resumo
As cardiopatias congênitas são as doenças congênitas de maior prevalência. O diagnóstico precoce, especialmente quando há repercussão fetal e neonatal, possibilita o aconselhamento familiar e a programação do parto em unidades adequadas para o atendimento de recém-nascidos que necessitam de manuseio de alta complexidade. A metodologia deste estudo consistiu em uma revisão da literatura, selecionando artigos relevantes sobre as principais cardiopatias congênitas com repercussão fetal e neonatal. O objetivo desta revisão é apresentar, de forma resumida, as cardiopatias mais importantes com repercussão fetal e neonatal.
Palavras-chave: Cardiopatias congênitas. Coração fetal. Recém-nascidos. Terapia intensiva neonatal.
Abstract
Congenital heart diseases are the most prevalent congenital disorders. Early diagnosis, especially when there are fetal and neonatal repercussions, enables family counseling and the planning of delivery in units equipped to handle newborns requiring high-complexity care. The study methodology comprised a literature review, selecting relevant articles on the main congenital heart diseases with fetal and neonatal repercussions. This review aims to present, in a summarized manner, the most important congenital heart diseases with fetal and neonatal repercussions.
Keywords: Congenital heart disease. Fetal heart. Neonate. Neonatal intensive care.
INTRODUÇÃO
Esta revisão enfoca as atualizações sobre o estado atual do diagnóstico e tratamento da doença cardíaca fetal. Um dos grandes objetivos do diagnóstico pré-natal é a detecção das cardiopatias congênitas (CC) graves, cujo prognóstico depende do planejamento do parto em centro de referência especializado.1-3 A taxa de detecção pré-natal para cardiopatia congênita está aumentando, mas apesar de a triagem anatômica pré-natal ter aumentado no Brasil, proporção significativa de cardiopatia congênita continua sem ser detectada antes do nascimento, uma vez que ocorrem em grupos de baixo risco e não são detectados na ultrassonografia pré-natal.4,5
Atualmente, vários estudos comprovam significativa diminuição de mortalidade e melhores resultados pré e pós-operatória nas CC quando diagnosticadas na vida fetal.6-8 Por esse motivo, esforços e políticas públicas devem dar continuidade à propagação de um padrão melhor e mais uniforme de rastreamento pré-natal das CC pelo ultrassom obstétrico, uma vez que a realização da ecocardiografia fetal especializada em todas as gestantes é utópica.9-11
CARDIOPATIAS CONGÊNITAS OBSTRUTIVAS DO CORAÇÃO ESQUERDO
Estenose aórtica
Anatomia e incidência
A estenose aórtica (EA) é definida como um estreitamento anatômico no nível da válvula aórtica. É, portanto, obstrução da via de saída do ventrículo esquerdo (VE), que em alguns casos pode levar a falha secundária do desenvolvimento do coração esquerdo e progressão final para SCEH. Esta evolução grave é devido a uma possível disfunção do VE, dano do miocárdio e redução do fluxo no coração esquerdo. Dependendo do local da obstrução, podemos classificar a EA como: valvar, subvalvar (membranosa ou tubular) e supravalvar.
A ecocardiografia fetal (EF) permite a avaliar a gravidade da obstrução, sendo difícil o diagnóstico da forma leve da EA no período neonatal. A EA ocorre em 0,2-0,5 por 1.000 nascidos vivos12,13 e representa cerca de 3-6% das todas as cardiopatias congênitas (CC). Pode estar associada a válvula aórtica bicúspide, síndrome de Shone, síndrome do coração esquerdo hipoplásico (SCEH), anomalias da válvula mitral, fibroelastose do ventrículo esquerdo (VE) e comunicação interventricular. A literatura mostra associação das diversas formas de EA com síndromes genéticas como a trissomia do 18, 13 e na trissomia parcial do 22 e em especial a síndrome de Williams-Bauren com a EA supravalvar.14,15
Diagnóstico e manejo pré-natal
A ecocardiografia fetal (EF) e suas inúmeras possibilidades diagnósticas permite avaliar a gravidade da EA, as lesões cardíacas associadas. No entanto, ainda é um grande desafio a diferenciação dos casos com potencial de progressão da obstrução durante a gravidez. Atualmente, a EF precoce realizada a partir 13ª semana de idade gestacional (IG) com a possibilidade de visualização das vias de saída, por via transvaginal ou abdominal, possibilita identificar precocemente esta complexa CC.(16)
Nos planos de quatro câmaras (4C), cinco câmaras (5C) e de via de saída do VE, identificamos a valva aórtica (VAo) espessada, mobilidade reduzida e sem abertura sistólica visível. A VAo, aorta ascendente pode ter calibre reduzidos e até hipoplasia significativa. Nesses planos, identificamos a EA supravalvar e subvalvar, achados menos comuns. Utilizamos o MMode para a mensuração dos ventrículos e avaliação da função ventricular, O mapeamento color/ doppler na aorta ascendente e na válvula mitral, permite mostra a velocidade o padrão turbulento do fluxo sistólico (velocidade sistólica máximo >100cm /s). No caso de dupla lesão aórtica, podemos identificar o fluxo diastólico na cavidade do VE. A identificação da direção do fluxo no nível atrial através do forame oval (FO) ou comunicação interatrial (CIA), quando esquerdo-direito ou bidirecional, serve como auxiliar no escore de gravidade. da EA. Podemos suspeitar de fibroelastose, quando ecogenicidade na cavidade do VE é semelhante ao encontrado no pericárdio e a maioria associa-se a hipocontratilidade.
Os planos dos três vasos (3VT) e três vasos com traqueia (3VT) com estudo do mapeamento color /doppler permite medir a aorta transversa, istmo, ducto arterioso além de verificar a direção do fluxo sanguíneo (anterógrado ou retrógrado), principalmente na aorta transversa.
O avanço do cateterismo cardíaco pediátrico intervencionista e o surgimento da valvoplastia aórtica intraútero com balão permitiu a abordagem da EA principalmente, nas formas evolutivas, visando a prevenção de possível desenvolvimento para a SHCE.17,18 A Tabela 1 mostra os principais critérios ecocardiográficos para a suspeição da EA evolutiva.
Podemos citar como critérios importantes de exclusão para intervenção fetal a presença de hipoplasia ventricular esquerda (volume diastólico final ou escore z <-2 no eixo longo), hipoplasia da válvula mitral (escore z <-2), pressão baixa do VE (<30 mmHg avaliada por regurgitação mitral ou velocidade do jato de estenose aórtica) pois esses fatores são fortes preditores de circulação de ventrículo único.
Manejo pós-natal
Após o nascimento, aproximadamente 10-15% dos recém-nascidos portadores de EA irão necessitar prostaglandina e/ou uma intervenção hemodinâmica ou cirúrgica, para manter a perfusão sistêmica.19 Deve ser realizada uma avaliação minuciosa, para determinar se é indicada a manutenção de uma circulação biventricular ou a paliação univentricular, semelhante aos casos da SCEH.
Coarctação da aorta
Anatomia e incidência
A coarctação da aorta (CoAo) é um estreitamento do arco aórtico. Ocorre mais comumente na região do istmo da aorta (entre a artéria subclávia esquerda e o ducto arterial). Fetos com coarctação grave da aorta dependem de um ductus arterioso patente (DAP) para preservar o fluxo sanguíneo sistêmico e a perfusão tecidual. A CoAo pode estar presente com um estreitamento isolado do istmo aórtico ou com hipoplasia tubular do arco aórtico (arco transverso e istmo, o espectro de gravidade é variável, que vai desde o fluxo sanguíneo sistêmico canal-dependente, até pacientes com pequenas dimensões de arco limítrofes também têm dependência do DAP, sendo esse diagnóstico um desafio para o especialista mesmo após o nascimento.20
A CoAo é encontrada em 6% a 8% dos pacientes com cardiopatia congênita. É frequente na monossomia do X (síndrome de Turner). Algumas das malformações cardíacas e extra cardíacas que podem ser encontradas em associação à CoAo são: persistência da veia cava superior esquerda; complexo de Shone (associação de várias lesões obstrutivas do coração esquerdo); valva aórtica bicúspide/hipoplásica; membrana subaórtica, CIV, displasia/hipoplasia da válvula mitral; hipoplasia do VE; hipoplasia do arco aórtico e a hérnia diafragmática. (21)
Diagnóstico e manejo pré-natal
O diagnóstico pré-natal da CoAo é um desafio conhecido como "tendão de Aquiles" do cardiologista fetal, pois o canal arterial no feto é grande, dificultando essa avaliação. Exceto nos casos em que o istmo aórtico é hipoplásico, esse diagnóstico é de extrema dificuldade em fetos com alto índice de falso-positivos e falso-negativos. A sensibilidade do diagnóstico fetal de CoAo varia de 50% a 72%, e o diagnóstico tardio de CoAo ainda é comum. Em alguns casos, mesmo após o nascimento, a ecocardiografia sequencial é recomendada para que o diagnóstico adequado seja feito (geralmente após o fechamento do canal arterial).20,22
O principal achado para a suspeição de CoAo na ecocardiografia fetal é a desproporção ventricular, que pode ser observada no plano de 4C (Figura 1A) e no eixo curto dos ventrículos. Nesses planos, o ventrículo esquerdo parece estreito quando comparado ao ventrículo direito. A predominância de cavidades direitas é fisiológica no terceiro trimestre, mas geralmente não deve exceder uma relação VD:VE >1,5.23, 24 No plano dos três vasos e traqueia (3VT), observa-se uma discrepância entre o tamanho dos grandes vasos, havendo diferença acentuada de tamanho entre a aorta ascendente (AAo) e a artéria pulmonar (AP) (Figura 1B). Uma proporção entre a AP e o AAo de ≥1,7 foi associada a CoAo, especialmente em fetos com mais de 28 semanas de gestação.
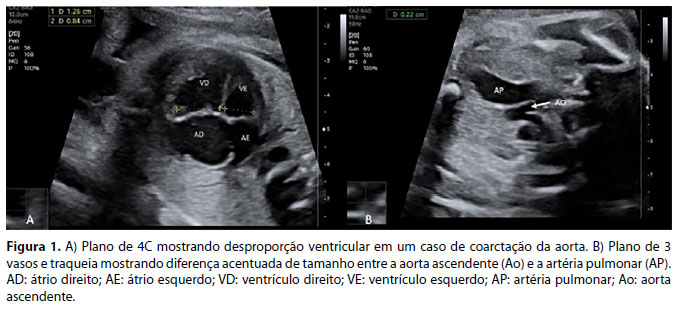
As diferenças de tamanho provavelmente são um reflexo de uma redistribuição do fluxo sanguíneo secundária ao aumento da resistência no trato de saída do ventrículo esquerdo com redução do fluxo sanguíneo através do AAo e um aumento compensatório no fluxo sanguíneo através do trato de saída direito através da AP e DAP em casos de CoAo. É importante avaliar o tamanho da valva mitral e o tamanho/morfologia da valva aórtica no bidimensional, color e doppler espectral. A quantificação das dimensões aórticas obtidas no nível do anel da valva aórtica, aorta ascendente (AAo), arco aórtico transversal, istmo e aorta descendente também devem ser realizadas. As medidas devem ser realizadas na parede interna do vaso em sua dimensão máxima obtida na sístole, quando a valva aórtica está aberta.
O exame doppler colorido e espectral é realizado no nível da valva aórtica, arco aórtico transversal, istmo, aorta descendente e arco ductal. A valva aórtica deve demonstrar o fluxo anterógrado e a integral do tempo de velocidade e a velocidade de pico registrada. O fluxo anterógrado ao doppler colorido em todo o arco aórtico, istmo e aorta descendente, bem como o desvio da direita para a esquerda através do DAP, devem ser demonstrados no corte sagital do arco aórtico. Um aumento de velocidade, fluxo contínuo ou reversão de fluxo através do istmo aórtico justificam uma avaliação mais detalhada pela possibilidade de CoA. A hipoplasia do arco transversal distal e istmo aórtico ocorrem quando o z escore é menor do que -2. A presença de uma prateleira posterior na região do istmo aórtico e deslocamento distal da artéria subclávia esquerda são características ecocardiográficas no eco fetal.
Uma meta-análise revelou que a hipoplasia do arco aórtico teve a maior sensibilidade (90%) no diagnóstico de coarctação, enquanto uma prateleira posterior teve a maior especificidade. O índice da artéria carótida-subclávia, que é a razão entre o diâmetro do arco aórtico ao nível da artéria subclávia esquerda, e a distância entre a artéria carótida esquerda e a artéria subclávia esquerda, é menor em pacientes com coarctação.21 Dependendo da idade gestacional no momento da suspeita inicial, o ecocardiograma fetal pode ser realizado a cada 4-6 semanas, com a última avaliação obtida após 30 semanas. No mínimo, uma avaliação de acompanhamento no terceiro trimestre é razoável para avaliar a progressão de anormalidades e detectar aquelas que podem não ter sido evidentes no exame inicial.
Um importante preditor ecocardiográfico para CoA é a progressão no grau de hipoplasia das dimensões do VE e da aorta durante as avaliações seriadas. A vigilância deve incluir avaliação do crescimento do coração esquerdo (VE, valva mitral, valva aórtica, arco aórtico) com atenção à quantificação detalhada das dimensões da aorta e avaliação pelo doppler do coração esquerdo e do arco aórtico. Isso ajudará o especialista a prever de forma mais razoável as expectativas pós-natais e se o uso de prostaglandina (PGE) seria ou não necessário no pós-natal.
Manejo pós-natal
Na CoAo crítica, a evolução neonatal precoce para choque cardiogênico está associada à disfunção ventricular grave após fechamento do canal arterial. A infusão de prostaglandina deve ser iniciada após o parto até que um ecocardiograma pós-natal possa ser realizado para avaliar a anatomia do coração esquerdo e o arco aórtico. A vigilância ativa deve ser realizada, incluindo a avaliação dos pulsos da extremidade superior direita e de membros inferiores. O procedimento cirúrgico mais comum para correção da coarctação da aorta no período neonatal é a ressecção e anastomose término-terminal. Este procedimento consiste na ressecção da coarctação e anastomose (conexão) da aorta proximal e distal.25, 26
Síndrome de hipoplasia do coração esquerdo (SHCE)
É um conjunto de anomalias cardíacas congênitas, que consiste em hipoplasia significativa ou ausência do ventrículo esquerdo e hipoplasia da aorta ascendente.27,28 Na sua forma mais grave, ocorre atresia das valvas aórtica e mitral e ausência de cavidade do ventrículo esquerdo funcional. Caracterizada por um subdesenvolvimento das estruturas cardíacas do lado esquerdo (valva mitral, ventrículo esquerdo, valva aórtica e arco aórtico), o lado esquerdo do coração é incapaz de suportar a circulação sistêmica. O sangue que retorna dos pulmões deve fluir através de um defeito do septo atrial para o átrio direito e, então, para o ventrículo direito.29,30 O ventrículo direito bombeia o sangue para a artéria pulmonar e o sangue atinge a circulação sistêmica através do ducto arterioso patente. Em resumo, há hipoplasia grave da cavidade ventricular esquerda e trato de saída, com um ventrículo direito dominante.30
Epidemiologia / Genética
A SHCE compreende 1,4-3,8% de todas as doenças cardíacas congênitas.30,31 Há evidências crescentes que sugerem uma etiologia genética, mas, até agora, nenhuma anormalidade genética específica foi associada à SHCE. Com um membro da família afetado, há cerca de 2-13,5% de risco de recorrência de outra doença cardíaca congênita na mesma família.
Anatomia / Classificação
A SHCE ocorre em um espectro que varia de atresia da valva aórtica com atresia mitral (AA/AM), atresia da valva aórtica com uma valva mitral patente com estenose (AA/MS) e estenose aórtica com uma valva mitral patente com estenose (AS/MS).31
Além disso, outras anormalidades congênitas podem estar presentes. As fístulas de câmara coronária são vistas em pacientes com atresia aórtica com uma valva mitral patente. Septo atrial intacto ou restritivo ocorre em 1% dos recém-nascidos com hipoplasia de cavidades esquerdas. Veia cava superior esquerda persistente ocorre em 15% e a drenagem venosa pulmonar anômala em 5-10%.31
Evolução pré-natal e apresentação clínica pós-natal
A SHCE geralmente é diagnosticada por ultrassom pré-natal, dado o avanço na ecocardiografia fetal. Esses pacientes necessitam prostaglandina (PGE) pós-natal para manter a patência ductal até que o procedimento híbrido ou a cirurgia paliativa estágio I possa ser realizada. Fetos com estenose aórtica crítica (que podem evoluir para variantes da SHCE) podem ser candidatos à valvuloplastia aórtica fetal. Da mesma forma, aqueles com septo atrial restritivo/intacto são candidatos à colocação de stent intra-atrial durante a vida fetal no período pós-natal imediato.
Crianças nascidas sem diagnóstico pré-natal podem ter manifestações clínicas variáveis. Aqueles com septo atrial restritivo/intacto desenvolvem cianose e desconforto respiratório no início, enquanto aqueles com uma comunicação atrial não restritiva podem parecer acianóticos inicialmente e vir ao atendimento médico com o fechamento do ducto arterial patente (DAP) e exibir sinais de má perfusão sistêmica, como desconforto respiratório, extremidades pálidas e frias, letargia etc.
Objetivos da ecocardiografia fetal
Em geral, a suspeita da SCEH é realizada pela ultrassonografia obstétrica pela imagem alterada das quatro câmaras do coração fetal. A ecocardiografia fetal confirma o diagnóstico e possibilita o detalhamento anatômico, fornecendo informações sobre mensurações do ventrículo esquerdo e a aorta. Essas medidas são plotadas em Z escores para idade gestacional ou idade gestacional acrescidas da biometria fetal, possibilitando a avaliar o grau de hipoplasia do VE e sua via de saída. Pode ocorrer evolução com restrição do forame oval e /ou canal arterial, piora da função do miocárdio do VD e disfunção valvar tricúspide, indicando a realização de exames de ecocardiografia fetal de seguimento. Essas informações são importantes para o planejamento cirúrgico pós-natal de crianças com SHCE.31,32
Complexo de Shone
O complexo de Shone é um raro defeito cardíaco congênito que consiste em múltiplas lesões cardíacas obstrutivas do lado esquerdo, incluindo: anel ou membrana mitral supravalvar, válvula mitral em paraquedas, membrana subaórtica e coarctação da aorta.33 A variante de Shone, ou síndrome incompleta de Shone, é mais comum e tipicamente diagnosticada quando apenas duas ou três obstruções do lado esquerdo estão presentes na hipoplasia do coração esquerdo (valva mitral, valva aórtica, arco aórtico e ventrículo esquerdo).
Uma membrana mitral supravalvar é caracterizada por uma membrana de tecido conjuntivo superior ao orifício da valva mitral, que causa restrição do fluxo sanguíneo através da valva. Uma valva mitral em paraquedas é tipicamente caracterizada por ligações cordais a um único músculo papilar, mais comumente o músculo papilar posteromedial. Isso resulta em um anel da valva mitral estreito. Os folhetos da valva mitral são frequentemente espessados e displásicos com mobilidade reduzida e restrição de fluxo.
A estenose subaórtica pode ser membranosa ou muscular com restrição do fluxo sanguíneo abaixo da valva aórtica e através do trato de saída do ventrículo esquerdo.1,2,34 Uma valva aórtica bicomissural é um achado comum em síndrome de Shone e normalmente resulta em estenose aórtica valvar. A coarctação da aorta é descrita como estreitamento do arco aórtico, tipicamente no nível do istmo aórtico. O arco transversal pode ser alongado e hipoplásico. Esse estreitamento dificulta a passagem do sangue, resultando em menos sangue sendo enviado para a parte inferior do corpo. O complexo Shone, quando todos os quatro defeitos estão presentes, é raro, e o termo "complexo de Shone é frequentemente usado para se referir aos vários locais de obstrução e hipoplasia do lado esquerdo do coração.33,35
Intervenção cardíaca fetal
A intervenção cardíaca fetal é altamente especializada e tecnicamente desafiadora durante a vida fetal. É reservada para fetos com doença cardíaca congênita complexa. Em vários fetos de alto risco, pode aliviar a disfunção ventricular e impedir que alguns fetos desenvolvam hipoplasia ventricular progressiva à medida que a gestação avança e alcançar uma circulação biventricular pós-natal com melhor sobrevida fetal.27
Os candidatos a intervenções cardíacas fetais estão restritos a: estenose crítica da valva aórtica com evolução para síndrome hipoplásica do coração esquerdo, atresia pulmonar com um septo ventricular integro e síndrome hipoplásica do coração direito em evolução, e SCEH com um septo atrial integro ou altamente restritivo.27,36 O procedimento intraútero nas obstruções pode promover o crescimento ventricular e vascular e melhorar os resultados pós-natais. As opções terapêuticas incluem: valvuloplastia aórtica fetal, valvuloplastia pulmonar ou criação de uma comunicação interatrial (via balão e/ou colocação de stent) em fetos com SHCE com septo atrial íntegro ou restritivo.27,36
Intervenções cardíacas fetais são realizadas nos fetos até 30 semanas. O sucesso técnico, o resultado biventricular e a sobrevivência fetal estão melhorando continuamente com o treinamento da equipe.27,36-38 A Figura 2 ilustra o procedimento de intervenção fetal.
CARDIOPATIAS CONGÊNITAS OBSTRUTIVAS DO CORAÇÃO DIREITO
Atresia pulmonar com septo íntegro (APSI) / Estenose pulmonar crítica (EPC)
Introdução e incidência
Atresia pulmonar refere-se à ausência de conexão entre o ventrículo direito e a artéria pulmonar. A estenose pulmonar crítica se refere ao paciente que mantém algum fluxo pulmonar anterógrado, no entanto, incapaz de manter o débito cardíaco.
Ambas são cardiopatias canal arterial e forâmen oval dependentes, e necessitam assistência com uso de prostaglandinas para manter a patência do canal arterial após o nascimento e, eventualmente, atriosseptostomia, para permitir fluxo interatrial livre após o nascimento.
A EP valvar é a terceira cardiopatia congênita mais comum, ocorrendo em torno de 5% dos nascidos vivos com cardiopatia39 a APSI é mais rara e ocorre em 3%.3
Anatomia ao ecocardiograma fetal
As características do ecocardiograma fetal são:
- No corte de quatro câmaras, o ventrículo direito se encontra normal, hipoplásico ou dilatado, com hipertrofia de suas paredes. Em presença de regurgitação tricúspide significativa, pode haver dilatação do átrio direito. O ventrículo esquerdo é normal, mas pode estar com diâmetro restrito, se o septo interventricular estiver muito desviado à esquerda pela alta pressão no ventrículo direito.40
- No corte das vias de saída, o anel pulmonar é pequeno ou hipoplásico. Pode haver uma valva pulmonar com anel hipoplásico e valva espessada com abertura em dome, ou a via de saída terminar em fundo cego muscular sem valva. A artéria pulmonar é pequena ou hipoplásica e geralmente tem tronco e ramos confluentes, diferente da atresia pulmonar com CIV.
- A medida do Z-score da valva tricúspide auxilia, quando há possibilidade de correção biventricular, pois se correlaciona bem com o tamanho do ventrículo direito.
- O doppler colorido mostra fluxo reverso, da aorta para a artéria pulmonar, pelo canal arterial e na EPc pode haver algum fluxo anterógrado pela valva pulmonar (Figura 3). A presença de regurgitação tricúspide em graus variáveis não é rara.
- Na APSI, o doppler colorido também deve avaliar a circulação coronariana, em busca de conexões desta com o ventrículo direito, tornando o fluxo coronariano dependente da pressão do ventrículo direito. Nestes casos a descompressão do ventrículo direito pode levar queda da perfusão coronariana, com consequente isquemia miocárdica e até infarto.41,42
- O doppler espectral auxilia na avaliação do fluxo pulmonar, demonstrando se há algum fluxo anterógrado pela valva pulmonar (EPc) ou fluxo pulmonar totalmente alimentado pelo canal arterial (APSI). A presença de regurgitação tricúspide e sua quantificação também são importantes, pois velocidade do fluxo regurgitante menor que 200cm/s sugere disfunção do ventrículo direito.42
Manejo pré-natal
A maioria dos pacientes com APSI e EPc não apresentam insuficiência cardíaca ao nascimento. O acompanhamento é clínico/ecocardiográfico, e na presença de EPc ou APSI com sinais de evolução para hipoplasia do ventrículo direito, pode-se lançar mão de valvoplastia pulmonar intrauterina por balão. Os principais critérios para indicação atualmente são: estenose pulmonar crítica ou atresia pulmonar com septo íntegro com via de saída membranosa (e não a muscular), com folhetos valvares identificáveis ou uma membrana; Z-score da valva tricúspide entre -2,5 e -4 pode auxiliar a indicação.43 Para esta intervenção, a relação risco x benefício deve ser muito bem avaliada, pois ainda há um número significativo de insucessos e perdas fetais.
Manuseio pós-natal
A principal característica do exame físico neonatal é a presença de cianose, que pode ser observada desde o primeiro dia de vida. A presença de impulsão do ventrículo esquerdo palpável com ausência de impulsão do direito, também pode ser observada 44
A vantagem do diagnóstico fetal é o pronto início do tratamento após o nascimento, antes do fechamento do canal arterial, o que levaria à hipoxemia crítica com acidose metabólica e risco de morte.
Os exames complementares neonatais mais importantes são:
Gasometria arterial: havendo diagnóstico fetal e sendo o canal arterial mantido aberto com prostaglandinas, a gasometria mostra hipoxemia, mas sem alteração do lactato.
Eletrocardiograma: Em geral o ritmo é sinusal, com eixo elétrico preservado. Recém-nascidos com ventrículo direito normal ou próximo ao tamanho normal podem apresentar hipertrofia ventricular direita. Se há hipoplasia, a força do ventrículo esquerdo predomina e hipertrofia ventricular esquerda se manifesta. Na presença de regurgitação tricúspide grave, aumento do átrio direito pode estar presente com onda "a" apiculada.44
Radiografia de tórax: a área cardíaca em geral é normal, assim como o fluxo pulmonar, se o canal arterial estiver pérvio. Na presença de regurgitação tricúspide significativa, o átrio direito pode estar aumentado e haver cardiomegalia. Como todo o débito cardíaco sai pela aorta, pode haver dilatação desta, e concavidade na região do hilo pulmonar, refletindo artérias pulmonares hipoplásicas. Mas, não há uma radiografia de tórax característica.44
Ecocardiograma: é o exame de eleição para avaliar tanto APSI com EPc. Mostra todos os detalhes da anatomia, principalmente a avaliação do ventrículo direito, seu tamanho e função, pensando no tipo de correção a ser oferecido. Realiza também a avaliação da circulação coronariana. Além disso, pode avaliar bem o trato de saída do ventrículo direito, tamanho do anel valvar pulmonar, presença de valva pulmonar com possibilidade de valvoplastia por balão ou via de saída muscularizada com infundíbulo hipertrofiado ou ausência de valva pulmonar que indiquem cirurgia.
O cateterismo cardíaco e a angiotomografia ficam reservados para casos especiais, com malformações adicionais de difícil diagnóstico, necessidade de melhor avaliação da circulação coronariana ou, no caso do cateterismo, como medida terapêutica.
Tratamento
Uma vez realizado o diagnóstico, a estratégia terapêutica depende do tamanho do ventrículo direito, do comportamento da circulação coronariana e do aspecto da via de saída.
Pacientes com EPc ou APSI com ventrículo direito viável (com três porções, sem hipoplasia grave e com circulação coronariana normal), anel valvar pulmonar com Z-score maior que -2, infundíbulo pérvio e estenose crítica ou atresia membranosa da valva pulmonar, o cateterismo intervencionista está indicado para valvuloplastia pulmonar por balão. Em se tratando de atresia pulmonar, o procedimento inclui perfuração da valva por radiofrequência, seguida de valvuloplastia.44 Algumas vezes, após o procedimento, a hipertrofia do infundíbulo causa obstrução significativa da via de saída impedindo o fluxo pulmonar; nestes casos, a realização de uma derivação sistêmico-pulmonar é temporariamente necessária, até que, com a abertura da valva, a obstrução diminua. Nestes casos, uma derivação sistêmico-pulmonar, seja a implantação de stent no canal arterial ou uma derivação do tipo Blalock-Taussig, por exemplo, podem ser a solução.
Se o ventrículo direito é inviável, a opção é a realização de uma derivação sistêmico pulmonar, mais comumente do tipo Blalock Taussig, para futura correção univentricular. Na presença de atresia pulmonar infundibular, se o ventrículo direito permite, uma alternativa é a valvotomia pulmonar (cirúrgica) com ampliação da via de saída do ventrículo direito. Nestes casos também pode ser necessária a manutenção de fluxo pulmonar por uma derivação sistêmico-pulmonar até que o fluxo pela via de saída do ventrículo direito seja pleno.44,45
O prognóstico depende principalmente das condições do ventrículo direito e da valva tricúspide, sendo mais reservado naqueles com circulação coronariana dependente do ventrículo direito, que são mais sujeitos a isquemia miocárdica e arritmias. O maior risco de complicações ocorre nos primeiros seis meses de vida. Após esse período, a taxa de complicações é menor, havendo boa qualidade de vida, independentemente do tipo de correção.45
REFERÊNCIAS
1. Hoffman JI, Kaplan S, Liberthson RR. Prevalence of congenital heart disease. Am Heart J. 2004;147(3):425-39.
2. Hoffman JI, Kaplan S. The incidence of congenital heart disease. J Am Coll Cardiol. 2002;39(12):1890-900.
3. Abuhamad A, Chaoui R. A practical guide to fetal echocardiography : normal and abnormal hearts. Fourth edition. ed. Philadelphia: Lippincott Williams & Wilkins; 2022. x, 782 pages p.
4. Araujo Junior E, Bravo-Valenzuela NJ, Peixoto AB. Perinatal Cardiology Part 2. Books B, editor2020.
5. Ximenes RS, Bravo-Valenzuela NJ, Pares DBS, Araujo Júnior E. The use of cardiac ultrasound imaging in first-trimester prenatal diagnosis of congenital heart diseases. J Clin Ultrasound. 2023;51(2):225-39.
6. Bonnet D, Coltri A, Butera G, Fermont L, Le Bidois J, Aggoun Y, et al. [Prenatal diagnosis of transposition of great vessels reduces neonatal morbidity and mortality]. Arch Mal Coeur Vaiss. 1999;92(5):637-40.
7. Tworetzky W, McElhinney DB, Reddy VM, Brook MM, Hanley FL, Silverman NH. Improved surgical outcome after fetal diagnosis of hypoplastic left heart syndrome. Circulation. 2001;103(9):1269-73.
8. Franklin O, Burch M, Manning N, Sleeman K, Gould S, Archer N. Prenatal diagnosis of coarctation of the aorta improves survival and reduces morbidity. Heart. 2002;87(1):67-9.
9. Kleinert S. Routine prenatal screening for congenital heart disease. Lancet. 1996;348(9031):836.
10. Wyllie J, Wren C, Hunter S. Screening for fetal cardiac malformations. Br Heart J. 1994;71(4 Suppl):20-7.
11. Donofrio MT, Moon-Grady AJ, Hornberger LK, Copel JA, Sklansky MS, Abuhamad A, et al. Diagnosis and treatment of fetal cardiac disease: a scientific statement from the American Heart Association. Circulation. 2014;129(21):2183-242.
12. Allan LD, Sharland GK, Milburn A, Lockhart SM, Groves AM, Anderson RH, et al. Prospective diagnosis of 1,006 consecutive cases of congenital heart disease in the fetus. J Am Coll Cardiol. 1994;23(6):1452-8.
13. Brick DH, Allan LD. Outcome of prenatally diagnosed congenital heart disease: an update. Pediatr Cardiol. 2002;23(4):449-53.
14. Nomura RM, Brizot MeL, Liao AW, Hernandez WR, Zugaib M. Conjoined twins and legal authorization for abortion. Rev Assoc Med Bras (1992). 2011;57(2):205-10.
15. Ewigman BG, Crane JP, Frigoletto FD, LeFevre ML, Bain RP, McNellis D. Effect of prenatal ultrasound screening on perinatal outcome. RADIUS Study Group. N Engl J Med. 1993;329(12):821-7.
16. Tegnander E, Eik-Nes SH, Johansen OJ, Linker DT. Prenatal detection of heart defects at the routine fetal examination at 18 weeks in a non-selected population. Ultrasound Obstet Gynecol. 1995;5(6):372-80.
17. Doyle EF. Pediatric cardiology : proceedings of the second world congress. New York: Springer-Verlag; 1986. l, 1340 p. p.
18. Garne E, Stoll C, Clementi M, Group E. Evaluation of prenatal diagnosis of congenital heart diseases by ultrasound: experience from 20 European registries. Ultrasound Obstet Gynecol. 2001;17(5):386-91.
19. Del Bianco A, Russo S, Lacerenza N, Rinaldi M, Rinaldi G, Nappi L, et al. Four chamber view plus three-vessel and trachea view for a complete evaluation of the fetal heart during the second trimester. J Perinat Med. 2006;34(4):309-12.
20. Hornberger LK, Eckersley LG. Aortic Coarctation: The Fetal Cardiologist's Achilles Heel. Circ Cardiovasc Imaging. 2021;14(7):e012877.
21. Familiari A, Morlando M, Khalil A, Sonesson SE, Scala C, Rizzo G, et al. Risk Factors for Coarctation of the Aorta on Prenatal Ultrasound: A Systematic Review and Meta-Analysis. Circulation. 2017;135(8):772-85.
22. Kailin JA, Santos AB, Yilmaz Furtun B, Sexson Tejtel SK, Lantin-Hermoso R. Isolated coarctation of the aorta in the fetus: A diagnostic challenge. Echocardiography. 2017;34(12):1768-75.
23. Mărginean C, Mărginean CO, Muntean I, Togănel R, Voidăzan S, Gozar L. The role of ventricular disproportion, aortic, and ductal isthmus ultrasound measurements for the diagnosis of fetal aortic coarctation, in the third trimester of pregnancy. Med Ultrason. 2015;17(4):475-81.
24. Durand I, Deverriere G, Thill C, Lety AS, Parrod C, David N, et al. Prenatal Detection of Coarctation of the Aorta in a Non-selected Population: A Prospective Analysis of 10 Years of Experience. Pediatr Cardiol. 2015;36(6):1248-54.
25. Lee MG, Brink J, Galati JC, Rakhra SS, Konstantinov IE, Cheung MM, et al. End-to-side repair for aortic arch lesions offers excellent chances to reach adulthood without reoperation. Ann Thorac Surg. 2014;98(4):1405-11.
26. Doshi AR, Chikkabyrappa S. Coarctation of Aorta in Children. Cureus. 2018;10(12):e3690.
27. Pedra SRFF, Zielinsky P, Binotto CN, Martins CN, Fonseca ESVB, Guimarães ICB, et al. Brazilian Fetal Cardiology Guidelines - 2019. Arq Bras Cardiol. 2019;112(5):600-48.
28. Allen HD, Shaddy RE, Penny DJ, Cetta F, Feltes TF. Moss and Adams' heart disease in infants, children, and adolescents : including the fetus and young adult. Ninth edition. ed. Philadelphia: Wolters Kluwer; 2016. 2 volumes (xxx, 1853, I-36 pages) p.
29. Morris C, Outcalt J, Menashe V. Síndrome hipoplásica do coração esquerdo: história natural em uma
população geograficamente definida. Pediatria. 1990;85(6).
30. Perolo A, Prandstraller D, Ghi T, Gargiulo G, Leone O, Bovicelli L, et al. Diagnosis and management of fetal cardiac anomalies: 10 years of experience at a single institution. Ultrasound Obstet Gynecol. 2001;18(6):615-8.
31. Bharati S, Lev M. The surgical anatomy of hypoplasia of aortic tract complex. J Thorac Cardiovasc Surg. 1984;88(1):97-101.
32. Seliem MA, Chin AJ, Norwood WI. Patterns of anomalous pulmonary venous connection/drainage in hypoplastic left heart syndrome: diagnostic role of doppler color flow mapping and surgical implications. J Am Coll Cardiol. 1992;19(1):135-41.
33. Lai WW. Echocardiography in pediatric and congenital heart disease : from fetus to adult. Chichester, West Sussex ; Hoboken, NJ: Wiley-Blackwell; 2009. xii, 796 p. p.
34. Boe NM, Rhee-Morris L, Towner D, Moon-Grady AJ. Prenatal diagnosis of omphalocele and left atrial isomerism (polysplenia) including complex congenital heart disease with ventricular noncompaction cardiomyopathy. J Ultrasound Med. 2008;27(7):1117-21.
35. Li YD, Meng H, Pang KJ, Li MZ, Xu N, Wang H, et al. Echocardiography in the diagnosis of Shone's complex and analysis of the causes for missed diagnosis and misdiagnosis. World J Clin Cases. 2022;10(11):3369-78.
36. Yuan SM, Humuruola G. Fetal cardiac interventions: clinical and experimental research. Postepy Kardiol Interwencyjnej. 2016;12(2):99-107.
37. Matsui H, Gardiner H. Fetal intervention for cardiac disease: the cutting edge of perinatal care. Semin Fetal Neonatal Med. 2007;12(6):482-9.
38. Tworetzky W, Wilkins-Haug L, Jennings RW, van der Velde ME, Marshall AC, Marx GR, et al. Balloon dilation of severe aortic stenosis in the fetus: potential for prevention of hypoplastic left heart syndrome: candidate selection, technique, and results of successful intervention. Circulation. 2004;110(15):2125-31.
39. Samánek M, Vorísková M. Congenital heart disease among 815,569 children born between 1980 and 1990 and their 15-year survival: a prospective Bohemia survival study. Pediatr Cardiol. 1999;20(6):411-7.
40. Eidem BW, Johnson JN, Lopez L, Cetta F. Echocardiography in pediatric and adult congenital heart disease. Third edition. ed. Philadelphia: Wolters Kluwer; 2021. pages cm p.
41. Berkley E, Chauhan SP, Abuhamad A, Committee SfM-FMP. Doppler assessment of the fetus with intrauterine growth restriction. Am J Obstet Gynecol. 2012;206(4):300-8.
42. Chikkabyrappa SM, Loomba RS, Tretter JT. Pulmonary Atresia With an Intact Ventricular Septum: Preoperative Physiology, Imaging, and Management. Semin Cardiothorac Vasc Anesth. 2018;22(3):245-55.
43. Yilmaz Furtun B, Morris SA. Catheter-Based Fetal Cardiac Interventions. J Cardiovasc Dev Dis. 2024;11(6).
44. Croti UA, Mattos SdS, Pinto Junior VC, Aiello VD, Moreira VdM, editors. Cardiologia e Cirurgia Cardiovascular Pediatrica. 2 ed. São Paulo, Brasil: Editora Roca; 2013.
45. Gottschalk I, Strizek B, Menzel T, Herberg U, Breuer J, Brockmeier K, et al. Severe Pulmonary Stenosis or Atresia with Intact Ventricular Septum in the Fetus: The Natural History. Fetal Diagn Ther. 2020;47(5):420-8.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()