Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 24(4) - Dezembro 2024
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Artigo Original
Cardiopatias congênitas com repercussão hemodinâmica pós-natal diagnosticadas pelo ecocardiograma fetal - Parte 2
Congenital heart diseases with postnatal hemodynamic repercussions diagnosed by fetal echocardiography - Part 2
Anna Esther Araujo e Silva1,2; Eliane Lucas1,3; Maria de Fátima Monteiro Pereira Leite1,4; Nathalie Jeanne Bravo-Valenzuela1,5; Talita Nolasco Loureiro1,6
DOI:10.31365/issn.2595-1769.v24i4p119-130
1. SOPERJ, Departamento de Cardiopediatria - Rio de Janeiro, Rio de Janeiro, Brasil - RJ - Brasil
2. Hospital Universitário Antônio Pedro, Universidade Federal Fluminense, Departamento de Pediatria - Niterói - RJ - Brasil
3. Faculdade de Medicina de Teresópolis (UNIFESO), Departamento de Pediatria - Teresópolis - RJ - Brasil
4. Instituto Nacional de Saúde da Mulher, da Criança e do Adolescente Fernandes Figueira - IFF- FIOCRUZ, Departamento de Pediatria - Rio de Janeiro - RJ - Brasil
5. IPPMG, Universidade Federal do Rio de Janeiro, Departamento de Pediatria - Rio de Janeiro - RJ - Brasil
6. Hospital Universitário Pedro Ernesto, Universidade Estadual do Rio de Janeiro, Departamento de Pediatria - Rio de Janeiro - RJ - Brasil
Endereço para correspondência:
Instituição: SOPERJ, Departamento de Cardiopediatria - Rio de Janeiro, Rio de Janeiro, Brasil - RJ - Brasil
Recebido em: 27/08/2024
Aprovado em: 20/10/2024
Resumo
Este artigo revisa algumas das doenças cardíacas congênitas mais significativas com repercussões hemodinâmicas durante os períodos fetal e neonatal. As anomalias conotruncais mais prevalentes incluem a transposição das grandes artérias e a tetralogia de Fallot. Juntamente com essas, a atresia pulmonar (com ou sem defeito do septo ventricular), a anomalia de Ebstein e a displasia tricúspide constituem as principais doenças cardíacas congênitas cianóticas que se beneficiam substancialmente do diagnóstico fetal. A detecção precoce facilita o aconselhamento parental e o planejamento do parto, reduzindo, assim, as taxas de morbidade e mortalidade. A metodologia é a mesma da parte 1, por levantamento bibliográfico de artigos relevantes das cardiopatias acima.
Palavras-chave: Cardiopatias Congênitas. Coração Fetal. Terapia Intensiva Neonatal. Recém-Nascido.
Abstract
This paper reviews some of the most significant congenital heart diseases with hemodynamic repercussions during the fetal and neonatal periods. The most prevalent conotruncal anomalies include transposition of the great arteries and tetralogy of Fallot. Alongside these, pulmonary atresia (with or without ventricular septal defect), Ebstein anomaly, and tricuspid dysplasia constitute the primary cyanotic congenital heart diseases that benefit substantially from fetal diagnosis. Early detection facilitates parental counseling and birth planning, thereby reducing morbidity and mortality rates. The same methodology of part 1 was used, consisting of a literature review and selecting relevant articles on the congenital heart disease described above.
Keywords: Heart Defects. Congenital. Fetal Heart. Intensive Care Units, Neonatal. Infant, Newborn, Diseases.
INTRODUÇÃO
As anomalias conotruncais, as cardiopatias congênitas (CC) com obstrução completa ao fluxo anterógrado pulmonar (atresia pulmonar com e sem comunicação interventricular), a doença de Ebstein e a displasia da valva tricúspide são CC com repercussão clínica pós-natal. A transposição das grandes artérias (TGA), a tetralogia de Fallot (TF) (anomalias conotruncais mais frequentes) e atresia pulmonar com e sem CIV são CC cianogênicas que necessitarão de manejo cirúrgico pós-natal para sobrevida desses pacientes. Nos casos mais graves de displasia Ebstein e não Ebstein da valva tricúspide, sinais de insuficiência cardíaca fetal, como ascite, derrame pleural e/ou pericárdico e hidropisia fetal, podem estar presentes.
Há alto risco de morte fetal com grande repercussão hemodinâmica fetal nos casos com shunt circular e o parto deve ser planejado com a equipe de atendimento pré-natal preparada para receber um paciente de alta complexidade, difícil manuseio clínico e com expectativa de cirurgia cardíaca. Assim, o diagnóstico pré-natal é crucial, contribuindo para diminuir a morbidade e a mortalidade relacionadas à essas cardiopatias.
Nesse cenário, é importante ressaltar que o rastreamento cardíaco ultrassonográfico pré-natal desempenha papel importante. Assim, as diretrizes internacionais e nacionais ampliaram os planos de imagem ultrassonográficos, que devem ser avaliados durante a ultrassonografia obstétrica morfológica, contemplando, além da imagem das quatro cavidades do coração fetal, a análise das imagens das vias de saída ventriculares (grandes artérias) e dos vasos do mediastino superior do feto. A suspeita da CC durante o rastreio ultrassonográfico obstétrico e a ecocardiografia fetal possibilitam o diagnóstico precoce, com impacto na redução da morbidade e mortalidade perinatal relacionada à malformação cardíaca.1,2
ANOMALIAS CONOTRUNCAIS
As anomalias conotruncais englobam malformações do infundíbulo (conus arteriosus) e das grandes artérias com conexões e alinhamentos ventrículo-arteriais anormais.3 Em relação à embriogênese, esse grupo de CC resulta de um desenvolvimento anormal da região conotruncal do coração fetal, sendo responsável por 25-30% das CC em bebês.4 As principais cardiopatias de grupo são: transposição das grandes artérias (TGA), tetralogia de Fallot (TF), dupla via de saída do ventrículo direito (DVSVD) e tronco arterial comum (truncus arteriosus).
1. Tetralogia de Fallot (TF)
Morfologia, incidência
TF é a forma mais comum de CC cianogênica, sendo responsável por 8 a 12% de todos os casos pós-natais com CC.5,6 Possui quatro características anatômicas: 1) comunicação interventricular (CIV); 2) dextroposição da aorta; 3) obstrução da via de saída do ventrículo direito (estenose pulmonar infundíbulo-valvar); e 4) hipertrofia do ventrículo direito.7 Essa tétrade de defeitos resulta de um desvio anterior do septo de saída (infundibular) em relação ao restante do septo ventricular durante a cardiogênese.8
Há um grande espectro de apresentações anatômicas e clínicas da TF de acordo com o grau de obstrução ao fluxo de saída do VD. Na forma clássica de TF, existe estenose pulmonar infundíbulo valvar cujo grau de obstrução ao fluxo pulmonar varia de leve a grave. Os recém-nascidos com a forma clássica de TOF e estenose pulmonar representam 80% da vida pós-natal. Assim é importante ficar claro que a forma clássica de TF não apresenta fluxo pulmonar dependente do canal arterial e, em geral, não requer abordagem cirúrgica durante o período neonatal.9,10 Entretanto, existe uma forma rara de TF com agenesia da valva pulmonar, em que predomina a insuficiência pulmonar, pois existem apenas resquícios ou ausência completa da valva pulmonar. Assim, as artérias pulmonares estarão aumentadas, com consequente compressão extrínseca das vias aéreas durante a vida fetal e prognóstico reservado.11, 12
Diagnóstico e manejo pré-natal da TF
A forma clássica de TF não apresenta repercussão hemodinâmica fetal. A avaliação dos planos de via de saída ventriculares e mediastino superior pela ultrassonografia/ecocardiografia fetal possibilita esse diagnóstico. A presença de uma grande comunicação interventricular (CIV) com aorta "cavalgando" essa comunicação deve chamar a atenção para a sua suspeita. A presença da obstrução ao fluxo de via de saída do VD, associada às demais alterações, confirma o diagnóstico de TF.1,2,12,13 (Figura 1).
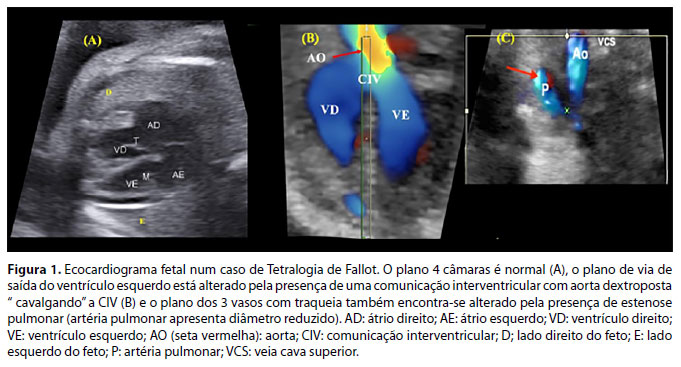
Durante o período fetal, recomenda-se a realização de exames de ecocardiografia de seguimento, pois poderá ocorrer evolução do grau de obstrução da via de saída do VD (= estenose pulmonar). O aconselhamento parental por equipe multidisciplinar é fundamental, explicando a cardiopatia à gestante e/ou ao casal, com suporte psicológico e planejamento do parto. Na TF clássica (forma mais comum), não há necessidade de parto em serviço com recursos de cirurgia cardíaca pediátrica, exceto quando a obstrução pulmonar for muito severa ou na forma com agenesia da valva pulmonar. A TF com agenesia da valva pulmonar (forma rara) cursa com mortalidade perinatal elevada. Em geral, dependendo da ressonância magnética fetal das vias aéreas e dos pulmões, o parto deverá ser planejado em um centro de referência com equipe de cardiologistas pediátricos e pediatras neonatologistas, cirurgia cardíaca pediátrica disponível e unidade de terapia intensiva neonatal com recursos de oxigenação por membrana extracorpórea (ECMO). A evolução com insuficiência cardíaca e respiratória está relacionada à insuficiência pulmonar, com consequente compressão extrínseca das vias aéreas pelas artérias pulmonares dilatadas.11,12,14
Manejo pós-natal da TF
Na TF forma clássica (sem estenose pulmonar importante), em geral, a correção cirúrgica será realizada durante o primeiro ano de vida. As manifestações clínicas dependerão do grau de obstrução da via de saída do VD; assim, se a estenose for leve, o recém-nascido (RN) poderá ser acianótico ou apresentar discreta cianose. Na ausculta cardíaca, em geral, revela a presença de sopro sistólico ejetivo de grau variável, mais audível em borda esternal esquerda alta. Quando a obstrução pulmonar for importante, há cianose evidente. Na radiografia de tórax, observaremos fluxo pulmonar reduzido (trama vascular reduzida). A oxigenioterapia é benéfica quando a SPO2 é inferior a 80% e desnecessária se superior a 90%. Em pacientes muito hipoxêmicos e/ou hemodinamicamente instáveis, a ventilação reduz o consumo de oxigênio ,"poupando" o trabalho respiratório, melhorando a perfusão tecidual. Nos casos de TF extrema (estenose pulmonar crítica), deve-se iniciar a PGE1 em infusão venosa contínua (por acesso central), na dose inicial de 0,01mcg/kg/min.(15) Essa dose poderá ser aumentada gradativamente (0,05 mcg/kg/min a cada 10 minutos até a dose de 0,1mcg/kg/min) até alcançar a melhora desejada. Irritabilidade, hipertermia e apneia são os efeitos adversos frequentes em doses elevadas.15, 16
O eletrocardiograma (ECG) demonstra eixo QRS+ 120o, padrão de hipertrofia do VD (rSR' ou Rs em V1). O ecocardiograma transtorácico confirma o diagnóstico e avalia o grau de estenose pulmonar (EP). Nas formas clássicas, maioria dos casos, realiza-se a correção cirúrgica ao longo do primeiro ano de vida, após período neonatal.17,18
A abordagem cirúrgica paliativa ainda no período neonatal (operação de Blalock-Taussig) será indicada apenas para casos de TF com estenose pulmonar crítica. Consiste em anastomose entre as artérias subclávia e pulmonar que aumenta o fluxo pulmonar e melhora a hipoxemia. Na correção total são realizados o fechamento da comunicação interventricular e ampliação da via de saída do VD. A forma de TF com agenesia da valva pulmonar poderá evoluir com insuficiência cardíaca congestiva, insuficiência respiratória e mortalidade perinatal elevada, sendo importante o planejamento do parto para manejo pós-natal inicial com recursos avançados de ventilação mecânica e oxigenação por membrana extracorpórea (ECMO) em unidade de terapia intensiva.11,14
2. Transposição das grandes artérias
Morfologia, incidência
A transposição das grandes artérias (TGA) é a segunda anomalia conotruncal mais frequente, correspondendo a 5-7% de todas as CC.19,20 Na TGA, a aorta origina-se no ventrículo anterior, que é o morfologicamente direito, e a artéria pulmonar no ventrículo morfologicamente esquerdo (posterior). Caracteriza-se, portanto, pela concordância atrioventricular (átrios conectam-se aos seus respectivos ventrículos) e discordância ventrículo-arterial (artérias transpostas), com um "arranjo" em paralelo das grandes artérias (Figura 2A).
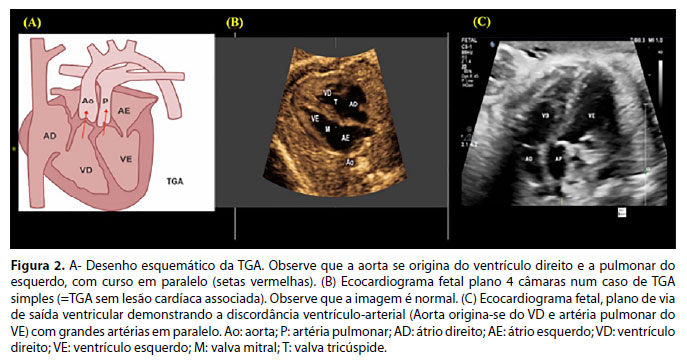
A TGA pode ser classificada como simples, quando não há anomalias cardíacas associadas, ou complexa, na qual as artérias transpostas coexistem com outras malformações cardíacas.
Apesar de mais frequente, a TGA simples ainda é uma das CC rotineiramente subdiagnosticada no período fetal.21,22
Diagnóstico e manejo pré-natal da TGA
Na TGA, a imagem ultrassonográfica das quatro câmaras do coração fetal é normal, exceto quando a TGA é complexa, como por exemplo, associada a uma comunicação interventricular (Figura 2B). As grandes artérias apresentam uma relação anormal: não se cruzam, estando dispostas em paralelo (Figura 2C). A avaliação das características das grandes artérias é fundamental para permitir o diagnóstico. A bifurcação da artéria que se origina do VE confirma esse diagnóstico. Para avaliar as características das grandes artérias, deve-se seguir as recomendações das diretrizes atuais que norteiam o rastreio cardíaco pré-natal.1,2,11 A ecocardiografia fetal poderá confirmar o diagnóstico de TGA e possibilitar o detalhamento anatômico, reduzindo a morbimortalidade e possibilitando o planejamento do parto em hospital com os recursos de UTI neonatal, hemodinâmica, cardiologia e cirurgia cardíaca pediátricas. Entretanto, o diagnóstico pré-natal da TGA ainda é um desafio.
Exames de seguimento buscam diagnosticar evolução com restrição do fluxo do forame oval e do canal arterial in utero. Nos fetos com forame oval restritivo, a atrioseptostomia (cateter balão/laser/stent) é um procedimento que poderá melhorar o prognóstico. É realizado entre 21 e 32-33 semanas de gestação, em centros de referência para intervenções em Medicina Fetal.23
Em relação ao fluxo do canal arterial fetal, a dieta rica em polifenóis (flavanoides ou não) e medicamentos com potencial anti-inflamatório (inclusive analgésicos como dipirona e paracetamol) oferecem risco para a constrição ductal no terceiro trimestre da gestação, por interferir na síntese das prostaglandinas. É importante orientar as gestantes no terceiro trimestre quanto a evitar alimentos ricos em polifenóis (≥ 30 mg polifenóis /100 g de alimento), objetivando um consumo < 125 mg de polifenóis por dia (classe II, nível A).(2, 24, 25)
Manejo pós-natal da TGA
A TGA é uma CC cianogênica com fluxo pulmonar dependente de alguma comunicação entre os átrios ou entre os ventrículos. A manutenção da patência do canal arterial também é muito importante para que o sangue oxigenado chegue até a aorta. Quando a mistura é inadequada (como na TGA simples com forame oval e canal arterial em fechamento), a cianose e a taquipneia aparecem no primeiro ou primeiros dias de vida. A cianose é persistente, não ocorrendo melhora significativa com oxigênio. Estes sintomas podem progredir rapidamente para acidose metabólica e morte se o tratamento adequado não for realizado. Ao exame físico, os pulsos periféricos e o precórdio geralmente são normais e pode não ocorrer sopro. Quando há comunicação interventricular associada (TGA com CIV), a cianose é menos intensa e os sintomas surgem mais tardiamente no período neonatal (insuficiência cardíaca).
Tipicamente, observaremos a radiografia de tórax: cardiomegalia (coração em forma de ovo deitado), mediastino estreito e aumento da circulação pulmonar. Na TGA simples, em geral, há desvio de eixo do QRS para a direita e hipertrofia ventricular direita (R em V1 e RS em V6) no ECG. A ecocardiografia transtorácica confirma o diagnóstico fetal da TGA e avalia a circulação pós-natal ou realiza o diagnóstico na ausência de diagnóstico pré-natal. Nos recém-nascidos com pouca mistura circulatória e cianose importante, a atriosseptostomia por cateter-balão (cateter de Rashkind) é um procedimento importante, a ser realizado nas primeiras 48-72 horas de vida. Nos casos com septo interatrial rígido, é realizada a atriosseptostomia cirúrgica (cirurgia de Blalock-Hanlon). Diante do diagnóstico de TGA, deve-se iniciar infusão contínua de prostaglandina para manter o canal arterial aberto, objetivando mistura entre as circulações até que a atriosseptostomia seja realizada. Esse procedimento geralmente melhora a saturação de oxigênio em 15-25% e se a mistura no nível atrial estiver adequada, a prostaglandina deve ser reduzida.
A cirurgia definitiva atualmente indicada para correção anatômica da TGA simples, sem lesões obstrutivas importantes do VE, é a cirurgia de Jatene ou switch arterial (troca arterial). Quando não é possível a correção anatômica, realiza-se a correção fisiológica (cirurgias de Mustard ou Senning), com inversão dos fluxos sistêmico e pulmonar para os ventrículos na região dos átrios (switch atrial).26
3. Dupla via de saída do VD e tronco arterial comum
Morfologia, fisiopatologia
A dupla via de saída do ventrículo direito (DVSVD) é uma cardiopatia congênita em que as duas grandes artérias se originam completamente, ou em grande parte, do ventrículo direito (VD). Considera-se que uma artéria pertence a um ventrículo quando mais de 50% de sua circunferência está conectada a esse ventrículo. Assim, tipicamente, existe uma comunicação interventricular (CIV) que permite a conexão do VD com as grandes artérias (Figura 3A).27,28 A fisiologia hemodinâmica pós-natal dependerá, principalmente, da relação da CIV com as grandes artérias e da presença ou não de obstruções ao fluxo de via saída ventricular.
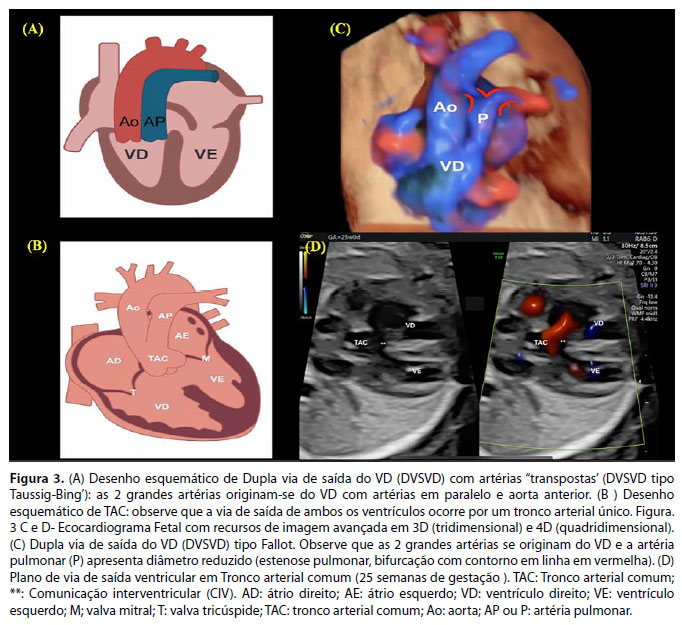
O tronco arterial comum (TAC), também denominado Truncus arteriosus, é uma cardiopatia congênita cianogênica em que o fluxo de ambos os ventrículos é ejetado para um vaso arterial único. Assim, em quase todos os casos, existe uma ampla comunicação interventricular que se localiza logo abaixo da valva truncal (Figura 3B). Do tronco arterial único (comum aos 2 ventrículos) originam-se as artérias coronárias, artérias pulmonares e a aorta ascendente. Na circulação pós-natal, ocorre cianose por mistura entre as circulações pulmonar e sistêmica (shunt misto). Com a queda progressiva da resistência vascular pulmonar após o nascimento, ocorrerá insuficiência cardíaca.28-30
Diagnóstico e manejo pré-natal
Para a suspeita dos diagnósticos de DVSVD pela ultrassonografia obstétrica durante o rastreio para malformação cardíaca no segundo trimestre, é importante que, além da avaliação do plano de quatro câmaras do coração fetal, sejam avaliados os planos de via de saída ventriculares e de mediastino superior. O ecocardiograma fetal possibilita o diagnóstico com detalhamento anatômico e seguimento pré-natal dessa CC (Figura 3C).28,31 Entretanto, durante a vida fetal, a diferenciação entre o TAC com outras anomalias conotruncais constitui um grande desafio, mesmo para experts em cardiologia fetal. (Figura 3D). A atresia pulmonar com comunicação interventricular (AP+CIV) é um importante diagnóstico diferencial, com apresentação clínica pós-natal diferente do TAC. Em ambas, o fluxo de saída dos ventrículos ocorre por apenas uma artéria (aorta na AP+CIV e vaso truncal no TAC). No TAC, a cianose é em geral mais discreta que na AP+CIV e com evolução para insuficiência cardíaca importante. A DVSVD e o TAC, em geral, se mantêm estáveis no período fetal, exceto nos casos de TAC com insuficiência importante da valva truncal em que ocorre evolução com insuficiência cardíaca fetal. O aconselhamento dos pais quanto ao prognóstico e opções de tratamento pós-natal é realizado durante o seguimento pré-natal.28,32
Manejo pós-natal
A ecocardiografia transtorácica, em geral, detalha a anatomia cardíaca e a circulação pós-natal. Na DVSVD com estenose pulmonar crítica ou atresia (DVSVD tipo Fallot), a cianose é a apresentação clínica principal. Nesses casos, deverá ser administrada a prostaglandina E1 para garantir a perfusão pulmonar. Nos casos de DVSVD sem obstrução ao fluxo de via de saída do VD (DVSVD tipo CIV), a apresentação clínica é com insuficiência cardíaca (IC), que ocorre, geralmente, após a terceira semana de vida (pela queda da resistência pulmonar pós-natal). Medidas anticongestivas devem ser instituídas até posterior conduta cirúrgica. Na DVSVD com vasos transpostos, o quadro clínico é de IC com cianose central. A ausculta de um sopro sistólico na borda esternal esquerda alta (estenose pulmonar), de um sopro diastólico em ponta, nos casos de hiperfluxo pulmonar, ou sopro de lesões associadas podem estar presentes. A radiografia de tórax varia com o espectro morfológico: área cardíaca normal quando associada ao hipofluxo pulmonar na DVSVD tipo Fallot; coração "ovo deitado" (DVSVD com artérias transpostas) e área cardíaca aumentada com hiperfluxo pulmonar (DVSVD tipo CIV). No período neonatal, poderão ser indicados procedimentos cirúrgicos paliativos, como atriosseptostomia (DVSVD com artérias transpostas com comunicação interatrial restritiva), cirurgia de BT modificado para os casos com cianose importante (hipofluxo pulmonar) e ainda a bandagem da artéria pulmonar para casos com IC neonatal (hiperfluxo pulmonar).26,31
No TAC, a principal manifestação clínica inicial no período neonatal será a cianose e, posteriormente, sinais de IC, que ocorrerão com a queda da resistência vascular pulmonar. A ausculta cardíaca será de um sopro diastólico de grau variável, dependendo do grau de regurgitação da valva truncal e uma 2ª bulha única pela inexistência do componente pulmonar. A radiografia de tórax demonstrará aumento da área cardíaca com mediastino superior alargado. O eletrocardiograma nos casos de TAC com hiperfluxo, mostrará sinais de aumento do átrio esquerdo e ventrículos. A ecocardiografia pós-natal possibilita o diagnóstico de TAC. Nos casos de anatomia complexa, a ressonância cardíaca magnética ou a angiotomografia do coração e seus vasos auxiliam na análise de detalhes anatômicos, entretanto, o risco-benefício pela anestesia para o procedimento devem ser considerados. A terapêutica clínica inicial para IC com medidas anticongestivas deverá ser iniciada pelo pediatra/neonatologista. A correção cirúrgica do TAC deverá ser indicada nos dois primeiros meses de vida (fechamento da CIV com conexão da neo-aorta ao ventrículo esquerdo e do VD ao tronco pulmonar).26
4. Atresia pulmonar com CIV (APCIV)
Considerada por algum tempo uma forma de tetralogia de tetralogia de Fallot extrema, a APCIV consiste em obstrução grave da via de saída do ventrículo direito acompanhada de CIV, no entanto, sua principal característica é a variabilidade do fluxo arterial pulmonar, que pode se apresentar desde a configuração com tronco e ramos pulmonares confluentes e levemente diminuição do seu calibre, até total ausência de artérias pulmonares verdadeiras, sendo todo fluxo pulmonar ofertado através de vasos colaterais sistêmico-pulmonares.33
É considerada uma doença rara, com prevalência de 10 a cada 100.000 nascidos vivos e corresponde a 2% das cardiopatias congênitas.34 Existe associação com deleção do 22q11, CATCH 22 e diabetes materno.35
As características do ecocardiograma fetal são:28
- O corte de quatro câmaras pode ser totalmente normal, mostrando apenas desvio do eixo.
- A comunicação interventricular quando ampla pode ser visualizada no corte de quatro câmaras, mas se torna especialmente aparente a partir do corte da via de saída do ventrículo esquerdo. A aorta cavalga o septo e recebe todo o débito de ambos os ventrículos
- No corte dos três vasos, a artéria pulmonar se mostra menor que a aorta, podendo ser muito hipoplásica, somente com ramos confluentes sem a visualização do tronco ou totalmente ausente
- A aorta habitualmente é dilatada, pois recebe todo o débito cardíaco combinado (VE e VD).
- No doppler colorido, na presença de artérias pulmonares verdadeiras, seu fluxo provém do canal arterial (Figura 4D). Quando estas estão ausentes, colaterais sistêmico-pulmonares, geralmente saindo da aorta descendente, são a única fonte de fluxo pulmonar. Artérias pulmonares verdadeiras também podem coexistir com colaterais.
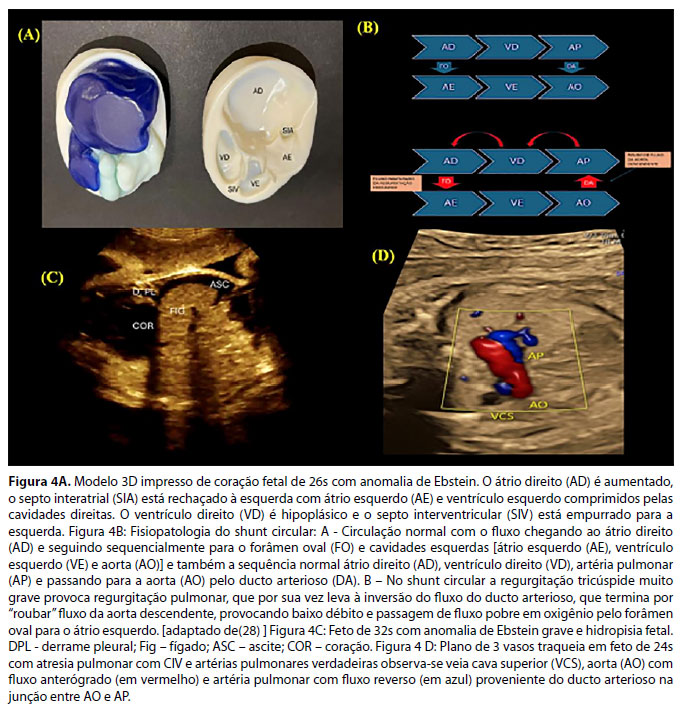
- A avaliação do corte sagital dos arcos aórtico e ductal, tanto em escala de cinza como no Doppler colorido, é uma excelente forma de individualização de pulmonares verdadeiras e colaterais.
O quadro clínico ao nascimento vai depender basicamente do formato da circulação pulmonar, que pode ser classificada da seguinte forma:36
a) todo fluxo pulmonar se origina das artérias pulmonares principais alimentadas pelo canal;
b) fluxo pulmonar se origina de artérias pulmonares principais, alimentadas pelo canal arterial, e de colaterais sistêmico-pulmonares, sendo suprido de ambas as formas;
c) todo fluxo pulmonar tem origem exclusivamente por colaterais sistêmico-pulmonares, com hipoplasia grave das artérias pulmonares principais e agenesia do ducto arterioso.
Assim, recém-nascidos com fluxo pulmonar exclusivamente por artérias pulmonares verdadeiras são dependentes do canal arterial e podem apresentar grave hipoxemia com seu fechamento. Já aqueles com circulação exclusivamente por colaterais não são dependentes do ducto arterioso, e o grau de hipoxemia ou de hiperfluxo vai depender da quantidade e qualidade das artérias colaterais. Naqueles em que a circulação é mista, manter o canal arterial pérvio ajuda no crescimento das artérias pulmonares verdadeiras, o que pode melhorar o prognóstico destes pacientes.37
Os principais exames complementares são:
Gasometria arterial: quanto maior o fluxo pulmonar, seja por colaterais ou pulmonares verdadeiras com canal patente, mais próxima do normal é a saturação de oxigênio e menor o risco de aumento do lactato. Nos primeiros dias, a saturação de oxigênio pode ser menor pela presença de resistência pulmonar elevada, mas tende a melhorar com a queda desta.
Eletrocardiograma: geralmente o ritmo é sinusal. O predomínio das cavidades ventriculares pode variar de acordo com o fluxo pulmonar: se baixo, as forças do ventrículo direito se sobrepõem, se o fluxo é aumentado, o aumento do retorno venoso pulmonar ao átrio e ventrículo esquerdos pode ser demonstrado pelos sinais de sobrecarga do ventrículo esquerdo.38
Radiografia de tórax: seu aspecto também é determinado pelo fluxo pulmonar. Quando este é equilibrado, a área cardíaca é normal, assim como a trama vascular pulmonar. Se há muitas colaterais, com a queda da resistência vascular pulmonar pode haver hiperfluxo e até congestão, além de aumento do índice cardiotorácico por aumento das cavidades esquerdas. Se as colaterais são pequenas, ou o fluxo através do canal arterial é restritivo, pode haver baixo fluxo pulmonar com área cardíaca normal.38
Ecocardiograma: é o método mais efetivo para diagnóstico, mostra a CIV, a função biventricular, mostra a atresia pulmonar e avalia a região infundibular. Em presença de pulmonares principais, pode visualizá-las, mas não consegue realizar a determinação exata da fonte do fluxo pulmonar quando há somente colaterais ou uma circulação mista.39
Angiotomografia e ressonância magnética: ambos são exames não invasivos que permitem identificar artérias pulmonares verdadeiras, mesmo que hipoplásicas e auxiliam na identificação de colaterais sistêmico-pulmonares, suas origens e trajetos.39
Cateterismo cardíaco: tem função diagnóstica e terapêutica.
Tratamento
Recém-nascidos com APCIV devem ser encaminhados à unidade de terapia intensiva ao nascimento, na dúvida sobre a presença de artérias pulmonares verdadeiras, até que sua existência seja comprovada ou afastada, deve ser iniciada a infusão de prostaglandinas. Na ausência de pulmonares verdadeiras com agenesia do ducto arterioso, não há indicação de tratamento medicamentoso para o fluxo pulmonar.
O objetivo final do tratamento é, sempre que possível, fechar a CIV, reconstruir a via de saída do ventrículo direito e recuperar o fluxo das artérias pulmonares principais irrigando o maior número possível de seguimentos pulmonares, fornecendo aos pulmões uma fonte única de fluxo com pressão adequada para evitar hipertensão pulmonar. Nos casos com múltiplas fontes de fluxo pulmonar, a unifocalização deve ser realizada sempre que possível.37,38 Esses procedimentos são avaliados caso a caso e realizados em etapa única ou de forma escalonada.
O prognóstico depende basicamente da possibilidade de manter o fluxo pulmonar saindo da cavidade ventricular direita, com pressão adequada. Desta forma, a presença de artérias pulmonares verdadeiras melhora o prognóstico, enquanto a presença de múltiplas colaterais com fluxo sistêmico-pulmonar com limitada capacidade de unifocalização têm pior evolução.
ANOMALIA DE EBSTEIN (AEB) E DISPLASIA TRICÚSPIDE (DT)
São as cardiopatias congênitas mais comuns da valva tricúspide. Na anomalia de Ebstein, ocorre um defeito na rotação e delaminação da valva tricúspide, em que os folhetos inferior e septal permanecem aderidos ao endocárdio do ventrículo direito, transformando esta porção do ventrículo direito em cavidade atrial direita, o que é chamado "atrialização".28 Também ocorre uma rotação destes folhetos, que passam a apontar para a via de saída do ventrículo direito. Como consequência, há regurgitação valvar tricúspide e obstrução do fluxo da via de saída do ventrículo direito, ambos em grau variável.40 Embora o folheto anterior se mantenha geralmente na posição habitual, ele também não é normal. A "atrialização" do ventrículo direito leva à insuficiência ventricular direita, em que, nos casos mais graves, o ventrículo direito não consegue gerar pressão suficiente para abrir a valva pulmonar, provocando atresia pulmonar funcional.
Na displasia da valva tricúspide, há displasia valvar associada a regurgitação, mas não há alteração na implantação ou na delaminação dos folhetos valvares, estando estes na sua posição normal.40 A frequência no período neonatal é de 1 a 10.000-50.000 nascidos vivos, e a prevalência pré-natal muito maior, entre 3 e 7%, demonstrando a gravidade da doença no período fetal. Apesar da evolução da tecnologia, a mortalidade neonatal, especialmente na anomalia de Ebstein grave, varia entre 27,4 e 50% em centros de excelência.41
O ecocardiograma fetal é diagnóstico, e mostra:28,40
- Aumento do átrio direito (algumas vezes é gigante e comprime os pulmões), ventrículo direito pequeno (ou hipoplásico) na AEb, podendo ser normal na DT. Átrio e ventrículo esquerdos podem ser comprimidos pelo átrio direito, podendo haver disfunção do ventrículo esquerdo. (Figura 1A)
- Doppler colorido: regurgitação tricúspide de graus variáveis. Fluxo de artéria pulmonar pode ser normal ou a artéria pulmonar pode ser alimentada por fluxo reverso pelo ducto arterioso. Eventualmente, observa-se fluxo bidirecional através do septo interventricular por uma comunicação interventricular (CIV).
- Regurgitação pela valva pulmonar evidencia a presença de shunt circular, em que o fluxo do ducto arterioso retorna pela artéria pulmonar e volta ao átrio direito pela regurgitação tricúspide, e volta ao lado esquerdo do coração pelo forâmen oval. Sua presença é fator de risco para morte fetal, pois rouba fluxo da aorta descendente e causa baixo débito e acidose metabólica (Figura 1 B). Nesses pacientes, em casos selecionados, pode ser necessário fechamento parcial do ducto arterioso para diminuir o roubo de fluxo da aorta descendente e assim, diminuir o baixo débito e evitar a morte fetal.41
- No doppler espectral, há regurgitação da valva tricúspide, em geral grave, com velocidades acima de 200cm/s. Baixas velocidades de fluxo ocorrem na disfunção do ventrículo direito.28
- No corte das vias de saída, a artéria pulmonar pode mostrar-se pequena, até hipoplásica, devido ao baixo débito pulmonar. Pode haver atresia pulmonar funcional ou verdadeira.28
- A avaliação do ritmo cardíaco é fundamental, já que existe associação entre AEb e vias de condução anômala e, também, a presença de um átrio direito gigante pode levar a arritmias atriais como fibrilação e flutter atrial.41
- Nos casos mais graves, sinais de insuficiência cardíaca fetal como ascite, derrame pleural e/ou pericárdico e hidropisia fetal podem estar presentes. Há alto risco de morte fetal (Figura 1C).
Quando o ecocardiograma fetal apresenta características de gravidade extrema, como insuficiência cardíaca e shunt circular pré-natal, o nascimento deve ser planejado, com a equipe de atendimento pré-natal preparada para receber um paciente de alta complexidade, difícil manuseio clínico e com expectativa de cirurgia cardíaca.
O recém-nascido pode estar extremamente grave, com insuficiência cardíaca e deve ser encaminhado diretamente à unidade de terapia intensiva neonatal (UTIN), e eventualmente, pode necessitar de manobras de reanimação ainda na sala de parto (SP).
Na apresentação neonatal nos casos menos extremos, a cianose é um sinal comum, assim como o sopro de insuficiência tricúspide. Hepatomegalia é um sinal importante de disfunção ventricular direita.
Os exames complementares mais importantes são:
Gasometria arterial: no paciente grave, é comum encontrar acidose metabólica associada a hipoxemia.
Radiografia de tórax: aumento importante da área cardíaca especialmente às custas do átrio direito, inclusive com compressão e hipoplasia pulmonar. O fluxo pulmonar pode estar diminuído na atresia pulmonar e não há congestão pulmonar. Índice cardiotorácico maior que 0,65 é fator de mau prognóstico.42
Eletrocardiograma: o ritmo costuma ser sinusal com sinais de aumento de átrio direito. A síndrome de pré-excitação com conexões atrioventriculares acessórias não é rara.
Ecocardiograma: no período neonatal confirma o diagnóstico fetal, estabelece a gravidade, avalia as alterações morfológicas, a função ventricular esquerda e direita. Também avalia as condições dos fluxos pulmonar e do ducto arterioso.
Tratamento
Casos mais leves da doença devem ser mantidos sob supervisão clínica, enquanto há fechamento espontâneo do ducto arterioso, observando se há estabilidade hemodinâmica durante o fechamento. A queda da resistência vascular pulmonar melhora o fluxo através da valva pulmonar e o retorno venoso ao átrio esquerdo. Vasodilatadores pulmonares como oxigênio e óxido nítrico podem ser usados para auxiliar neste processo.38
O uso de prostaglandinas para manutenção da permeabilidade do canal arterial deve ser reservado àqueles pacientes com atresia pulmonar e instabilidade hemodinâmica, que necessitam cirurgia para manutenção da circulação pulmonar. Nos casos em que a atresia pulmonar é funcional, o uso deve ser restrito ao período crítico e sob muito cuidado, pois o canal arterial atua como uma fonte de aumento da resistência vascular pulmonar e piora a função do ventrículo direito, aumentando a regurgitação tricúspide e favorecendo a instalação de shunt circular, piorando o quadro clínico.41,42
Pacientes hemodinamicamente instáveis, em que não há melhora com a queda da resistência pulmonar, beneficiam-se da cirurgia de Starnes (oclusão da via de entrada do ventrículo direito com um retalho fenestrado), que é um tipo de correção univentricular. O procedimento fecha a via de entrada do ventrículo direito, deixando apenas uma fenestra para a passagem do fluxo, e não danifica a valva tricúspide. Atualmente, alguns cirurgiões acreditam que, em casos selecionados, a cirurgia de Starnes pode ser revertida mais tarde para correção biventricular.41
A cirurgia do cone, idealizada pelo cirurgião brasileiro José Pedro da Silva, reconstrói a valva tricúspide, permitindo uma correção biventricular, mas não é ideal para o período neonatal.40 Sempre que possível, o tratamento deve evitar a cirurgia no período neonatal.
Referências
1. International Society of Ultrasound in Obstetrics and Gynecology, Carvalho JS, Allan LD, Chaoui R, Copel JA, DeVore GR, et al. ISUOG Practice Guidelines (updated): sonographic screening examination of the fetal heart. Ultrasound Obstet Gynecol. 2013;41(3):348-59.
2. Pedra SRFF, Zielinsky P, Binotto CN, Martins CN, Fonseca ESVB, Guimarães ICB, et al. Brazilian Fetal Cardiology Guidelines - 2019. Arq Bras Cardiol. 2019;112(5):600-48.
3. Van Praagh R. Definition of Conotruncal Anomalies. In: Lacour-Gayet F, Bove EL, Hraska V, Morell VO, Spray TL, editors. Surgery of Conotruncal Anomalies. Cham: Springer International Publishing; 2016. p. 1-25.
4. Anderson RH, Mori S, Spicer DE, Brown NA, Mohun TJ. Development and Morphology of the Ventricular Outflow Tracts. World J Pediatr Congenit Heart Surg. 2016;7(5):561-77.
5. Shinebourne EA, Babu-Narayan SV, Carvalho JS. Tetralogy of Fallot: from fetus to adult. Heart. 2006;92(9):1353-9.
6. Keane JF, Lock JE, Fyler DC, Nadas AS. Nadas' pediatric cardiology. 2nd ed. Philadelphia: Saunders Elsevier; 2006. xv, 934 p., 8 p. of plates p.
7. Anderson RH, Weinberg PM. The clinical anatomy of tetralogy of fallot. Cardiol Young. 2005;15 Suppl 1:38-47.
8. Stefanovic S, Etchevers HC, Zaffran S. Outflow Tract Formation-Embryonic Origins of Conotruncal Congenital Heart Disease. J Cardiovasc Dev Dis. 2021;8(4).
9. Marantz P, Grinenco S, Pestchanker F, Meller CH, Izbizky G. Prenatal diagnosis of CHDs: a simple ultrasound prediction model to estimate the probability of the need for neonatal cardiac invasive therapy. Cardiol Young. 2016;26(2):347-53.
10. Araujo Junior E, Bravo-Valenzuela NJ, Peixoto AB. Perinatal Cardiology Part 2. Books B, editor2020.
11. Donofrio MT, Moon-Grady AJ, Hornberger LK, Copel JA, Sklansky MS, Abuhamad A, et al. Diagnosis and treatment of fetal cardiac disease: a scientific statement from the American Heart Association. Circulation. 2014;129(21):2183-242.
12. Bravo-Valenzuela NJ, Lucas E, Silva AEA, Farias CVB. Atlas de Ecocardiografia Fetal. 1 ed. Rio de Janeiro, Brasil2021.
13. Medicine AIoUi. AIUM practice guideline for the performance of obstetric ultrasound examinations. J Ultrasound Med. 2010;29(1):157-66.
14. Bravo-Valenzuela NJ, Peixoto AB, Araujo Júnior E. Prenatal diagnosis of congenital heart disease: A review of current knowledge. Indian Heart J. 2018;70(1):150-64.
15. Yaffe SJ, Aranda JV. Neonatal and pediatric pharmacology : therapeutic principles in practice. 4th ed. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2011. xxiii, 1042 p. p.
16. Cucerea M, Simon M, Moldovan E, Ungureanu M, Marian R, Suciu L. Congenital Heart Disease Requiring Maintenance of Ductus Arteriosus in Critically Ill Newborns Admitted at a Tertiary Neonatal Intensive Care Unit. J Crit Care Med (Targu Mures). 2016;2(4):185-91.
17. Steiner MB, Tang X, Gossett JM, Malik S, Prodhan P. Timing of complete repair of non-ductal-dependent tetralogy of Fallot and short-term postoperative outcomes, a multicenter analysis. J Thorac Cardiovasc Surg. 2014;147(4):1299-305.
18. Martins IF, Doles IC, Bravo-Valenzuela NJM, Santos AORD, Varella MSP. When is the Best Time for Corrective Surgery in Patients with Tetralogy of Fallot between 0 and 12 Months of Age? Braz J Cardiovasc Surg. 2018;33(5):505-10.
19. Hoffman JIe. The global burden of congenital heart disease. Cardiovasc J Afr. 2013;24(4):141-5.
20. Unolt M, Putotto C, Silvestri LM, Marino D, Scarabotti A, Valerio Massaccesi, et al. Transposition of great arteries: new insights into the pathogenesis. Front Pediatr. 2013;1:11.
21. Escobar-Diaz MC, Freud LR, Bueno A, Brown DW, Friedman KG, Schidlow D, et al. Prenatal diagnosis of transposition of the great arteries over a 20-year period: improved but imperfect. Ultrasound Obstet Gynecol. 2015;45(6):678-82.
22. van Velzen CL, Haak MC, Reijnders G, Rijlaarsdam ME, Bax CJ, Pajkrt E, et al. Prenatal detection of transposition of the great arteries reduces mortality and morbidity. Ultrasound Obstet Gynecol. 2015;45(3):320-5.
23. Jaeggi E, Renaud C, Ryan G, Chaturvedi R. Intrauterine therapy for structural congenital heart disease: Contemporary results and Canadian experience. Trends Cardiovasc Med. 2016;26(7):639-46.
24. Vian I, Zielinsky P, Zílio AM, Schaun MI, Brum C, Lampert KV, et al. Increase of prostaglandin E2 in the reversal of fetal ductal constriction after polyphenol restriction. Ultrasound Obstet Gynecol. 2018;52(5):617-22.
25. Hanson MA, Bardsley A, De-Regil LM, Moore SE, Oken E, Poston L, et al. The International Federation of Gynecology and Obstetrics (FIGO) recommendations on adolescent, preconception, and maternal nutrition: "Think Nutrition First". Int J Gynaecol Obstet. 2015;131 Suppl 4:S213-53.
26. Herdy GVH, Silva AEA, Lucas E, Bravo-Valenzuela NJ. Cardiologia Pediátrica Pratica Clinica. 1 ed. Rio de Janeiro, 2022.
27. Ebadi A, Spicer DE, Backer CL, Fricker FJ, Anderson RH. Double-outlet right ventricle revisited. J Thorac Cardiovasc Surg. 2017;154(2):598-604.
28. Abuhamad A, Chaoui R. A practical guide to fetal echocardiography: normal and abnormal hearts. Fourth edition. ed. Philadelphia: Lippincott Williams & Wilkins; 2022. x, 782 pages p.
29. Collett RW, Edwards JE. Persistent truncus arteriosus; a classification according to anatomic types. Surg Clin North Am. 1949;29(4):1245-70.
30. Van Praagh R, Van Praagh S. The anatomy of common aorticopulmonary trunk (truncus arteriosus communis) and its embryologic implications. A study of 57 necropsy cases. Am J Cardiol. 1965;16(3):406-25.
31. Yagel S, Silverman NH, Gembruch U. Fetal cardiology : embryology, genetics, physiology, echocardiographic evaluation, diagnosis, and perinatal management of cardiac diseases. Third edition. ed. Boca Raton, FL: Taylor & Francis Group, LLC; 2019.
32. Volpe P, Paladini D, Marasini M, Buonadonna AL, Russo MG, Caruso G, et al. Common arterial trunk in the fetus: characteristics, associations, and outcome in a multicentre series of 23 cases. Heart. 2003;89(12):1437-41.
33. Gindes L, Salem Y, Gasnier R, Raucher A, Tamir A, Assa S, et al. Prenatal diagnosis of major aortopulmonary collateral arteries (MAPCA) in fetuses with pulmonary atresia with ventricular septal defect and agenesis of ductus arteriosus. J Matern Fetal Neonatal Med. 2022;35(25):5400-8.
34. Zhou J, Zhou Q, Peng Q, Zhang R, Tang W, Zeng S. Fetal pulmonary atresia with ventricular septal defect: Features, associations, and outcome in fetuses with different pulmonary circulation supply types. Prenat Diagn. 2019;39(12):1047-53.
35. Soquet J, Barron DJ, d'Udekem Y. A Review of the Management of Pulmonary Atresia, Ventricular Septal Defect, and Major Aortopulmonary Collateral Arteries. Ann Thorac Surg. 2019;108(2):601-12.
36. Tchervenkov CI, Roy N. Congenital Heart Surgery Nomenclature and Database Project: pulmonary atresia-ventricular septal defect. Ann Thorac Surg. 2000;69(4 Suppl):S97-105.
37. Anwar S, Rockefeller T, Raptis DA, Woodard PK, Eghtesady P. 3D Printing Provides a Precise Approach in the Treatment of Tetralogy of Fallot, Pulmonary Atresia with Major Aortopulmonary Collateral Arteries. Curr Treat Options Cardiovasc Med. 2018;20(1):5.
38. Croti UA, Mattos SdS, Pinto Junior VC, Aiello VD, Moreira VdM, editors. Cardiologia e Cirurgia Cardiovascular Pediatrica. 2 ed. São Paulo, Brasil: Editora Roca; 2013.
39. Abdel Razek AAK, Al-Marsafawy H, Elmansy M. Imaging of Pulmonary Atresia With Ventricular Septal Defect. J Comput Assist Tomogr. 2019;43(6):906-11.
40. Lianza AC, Rodrigues ACT, Mercer-Rosa L, Vieira MLC, de Oliveira WAA, Afonso TR, et al. Right Ventricular Systolic Function After the Cone Procedure for Ebstein's Anomaly: Comparison Between Echocardiography and Cardiac Magnetic Resonance. Pediatr Cardiol. 2020;41(5):985-95.
41. Konstantinov IE, Chai P, Bacha E, Caldarone CA, Da Silva JP, Da Fonseca Da Silva L, et al. The American Association for Thoracic Surgery (AATS) 2024 expert consensus document: Management of neonates and infants with Ebstein anomaly. J Thorac Cardiovasc Surg. 2024;168(2):311-24.
42. Sainathan S, da Fonseca da Silva L, da Silva JP. Ebstein's anomaly: contemporary management strategies. J Thorac Dis. 2020;12(3):1161-73.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()