Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 22(1) - Março 2022
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Bruno Previdelli Coghi
- Beatriz Bettiol Nicoletti
- Bruna Sandy Pastre
- Bruna Isepão Barboza da-Silva
- Zumira Aparecida Carneiro
- Jacqueline Fonseca
- Laura Vagnini
- Regina Albuquerque
- Marcela Almeida
- Debora de Cassia Tomaz
- Fernanda Veiga Goes
- Tainá Regina Damaceno Silveira
- Ana Paula Andrade Hamad
- Fernanda Timm
- Charles Marques Lourenco
Relato de Caso
Apresentação neuropsiquiátrica da doença de Niemann-Pick tipo C: relato de caso e revisão da literatura
Neuropsychiatric presentation of Niemann-Pick type C disease: case report and literature review
Bruno Previdelli Coghi1; Beatriz Bettiol Nicoletti1; Bruna Sandy Pastre1; Bruna Isepão Barboza da-Silva1; Zumira Aparecida Carneiro1; Jacqueline Fonseca2; Laura Vagnini3; Regina Albuquerque4; Marcela Almeida1; Debora de Cassia Tomaz4; Fernanda Veiga Goes5; Tainá Regina Damaceno Silveira6; Ana Paula Andrade Hamad7; Fernanda Timm8; Charles Marques Lourenco1,4
DOI:10.31365/issn.2595-1769.v22i1p41-46
1. Centro Universitario Estacio de Ribeirao Preto, Genetica Clínica - Ribeirao Preto - SP - Brasil
2. Laboratório DLE, Bioquímica Genética - Rio de Janeiro - RJ - Brasil
3. CPDP - Centro Paulista de Diagnóstico, Pesquisa e Treinamento - Medicina Diagnóstica, Genética Clínica - Ribeirao Preto - SP - Brasil
4. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - Ribeirao Preto - SP - Brasil
5. Instituto Nacional de Saúde da Mulher, Criança e do adolescente Fernandes Figueira (IFF) - Fiocruz, Neuropediatria - Rio de Janeiro - RJ - Brasil
6. Centogene AG, Genética Molecular - Rostock - Mecklemburgo-Pomerânia Ocidental - Alemanha
7. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - Ribeirao Preto - SP - Brasil
8. Hospital de Clinicas de Porto Alegre, UFRGS, Bioquímica Genética - Porto Alegre - RS - Brasil
Endereço para correspondência:
Recebido em: 13/04/2021
Aprovado em: 04/06/2021
Instituição: Centro Universitario Estacio de Ribeirao Preto, Genetica Clínica - Ribeirao Preto - SP - Brasil
Resumo
INTRODUÇÃO: Niemann-Pick tipo C (NP-C) é uma condição genética hereditária autossômica recessiva, causada por mutações nos genes NPC1 e NPC2, ocasionando armazenamento lisossomal neurovisceral. Alteração lisossomal decorre de defeito no trânsito celular, com consequente acúmulo de glicoesfingolipídios e colesterol em diversos órgãos, progressivamente levando à morte celular prematura. Essa doença apresenta uma prevalência de 1:120.000, no qual cerca de 95% dos afetados exibem alteração no gene NPC1 e apenas 5% são decorrentes de mutação no gene NPC2. Pacientes com NP-C podem apresentar vários fenótipos, desde a forma neonato-infantil, com manifestações viscerais como organomegalia e colestase, até a forma juvenil-adulta, com degeneração neurológica progressiva.
RELATO DE CASO: Paciente feminino, 18 anos, encaminhada para investigação de quadro neuropsiquiátrico ("esquizofrenia-like" e paralisia do olhar conjugado vertical), associado a manifestações motoras (ataxia, leve distonia e disartria). Diante dos achados clínicos e exame físico, solicitou-se o teste de Filipin que se positivou, sendo necessário o sequenciamento genético para comprovação da doença. Após o diagnóstico, iniciou uso de terapia de redução de substrato (TRS) com Miglustate, evoluindo com estabilização clínica.
DISCUSSÃO: NP-C deve ser considerada como diagnóstico diferencial de diversas doenças com manifestações viscerais e neuropsiquiátricas progressivas, a fim de direcionar sua investigação. Sendo a NP-C potencialmente tratável e dado seu caráter progressivo, o diagnóstico precoce torna-se fundamental, não só para melhor prognóstico clínico da paciente, mas também para permitir aconselhamento genético familiar.
Palavras-chave: Doença de Niemann-Pick Tipo C. Proteína C1 de Niemann-Pick. Colestase. Espectro da Esquizofrenia e Outros Transtornos Psicóticos.
Abstract
INTRODUCTION: Niemann-Pick type C (NP-C) is an inherited autosomal recessive genetic disease, caused by mutations in the NPC1 and NPC2 gene, causing neurovisceral lysosomal storage. Lysosomal storage is consequence of cellular traffic defect that leads to glycosphingolipid and cholesterol accumulation in many organs, progressing to premature cell death. This disease has a prevalence of 1: 120,000, in which about 95% of patients with the syndrome exhibit alterations in the NPC1 gene and only 5% have mutations in NPC2 gene. Patients with NP-C disease may present several phenotypes, from the neonatal-infantile form with visceral manifestations such as organomegaly and cholestasis, to the juvenile-adult form, with progressive neurological degeneration.
CASE REPORT: 18-year-old female patient referred for investigation of neuropsychiatric disorder ("schizophrenia-like" and vertical supranuclear gaze palsy), associated with motor manifestations (ataxia, mild dystonia and dysarthria). According to the clinical findings and physical examination, Filipin test and genetic sequencing were requested, confirming NP-C. After diagnosis, patient was started on substrate reduction therapy (TRS) with Miglustat, showing clinical stabilization.
DISCUSSION: NP-C should be considered as a differential diagnosis of several diseases with progressive visceral and neuropsychiatric manifestations, in order to direct its investigation. Since NP-C is potentially treatable and given its progressive character, early diagnosis becomes essential, not only for better clinical prognosis of the patient, but also to allow family genetic counseling.
Keywords: Niemann-Pick Disease, Type C. Niemann-Pick C1 Protein. Cholestasis. Schizophrenia.
INTRODUÇÃO
A doença de Niemann-Pick (NP), que atinge recém-nascidos, crianças e adultos de ambos os sexos, ocorre quando há acúmulo de esfingomielina, sendo dividida em três subgrupos principais. O subtipo A (OMIM 257200) e B (OMIM 607616) são causados pela deficiência da enzima esfingomielinase ácida (ASMD) lisossomal. O subtipo A tem suas manifestações nos primeiros meses de vida, sendo usualmente fatal; no subtipo B, o aparecimento dos sintomas ocorre na infância, sem desenvolver disfunção neurológica progressiva. Os pacientes com essa forma da doença apresentam, portanto, uma expectativa de vida maior quando comparada aos pacientes afetados por NPA; já o subtipo C (NP-C), causado por duas proteínas NPC1 e NPC2, evidencia o defeito do transporte da célula do colesterol e os sintomas neurodegenerativos que podem acometer desde a fase infanto-juvenil até a vida adulta.1,2
A NP-C tem uma prevalência de 1:120.000 nascidos e pode ser subdividida, de acordo com sua alteração genética, em dois loci gênicos: NPC1, que codifica uma proteína estrutural presente na membrana lisossomal; e NPC2, responsável por codificar uma proteína solúvel com função de transportador de colesterol, localizada no interior do lisossomo. Mutações tanto no gene NPC1 quanto NPC2 levam ao acúmulo de colesterol não esterificado dentro do lisossomo.4,5 Mutações do gene NPC2 correspondem a 5% dos casos de NP-C, enquanto cerca de 95% dos pacientes com NP-C apresentam mutações no gene NPC1.1,4
A enzima lipase ácida lisossomal promove a "quebra" do LDL e origina o colesterol não esterificado; o NPC 2 absorve e o transfere para NPC 1 que, por sua vez, libera para o citoplasma, o qual codifica uma glicoproteína localizada na membrana dos endossomos (Figura 1). A interrupção desse tráfego ocasiona o acúmulo de colesterol e acarreta distúrbios hepáticos e neurológicos.
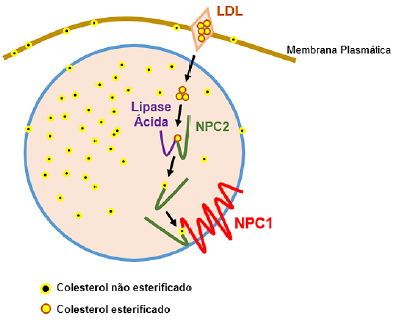
Figura 1. Ação dos genes NPC1 e NPC2 no metabolismo do LDL
Fonte: os autores (2021).
Pacientes portadores da doença NP-C mostram diversas manifestações clínicas, que são divididas em NP-C neonato-infantil e de fase juvenil-adulta,4,6 que abrange desordens neurológicas e hepáticas. Os sintomas viscerais na NP-C são mais frequentes e graves na fase neonato-infantil, quando hidropisia fetal, ascite fetal e icterícia colestática foram observadas.1,7
Já a forma juvenil-adulta tem predomínio de sintomas neurológicos, como deficiência intelectual, depressão, esquizofrenia e déficit de atenção; sintomas neurológicos, como a paralisia supranuclear do olhar vertical (vertical supranuclear gaze palsy, VSGP), ataxia, distonia, hipotonia, cataplexia, mioclonia e epilepsia.4,6-8 Vale ressaltar evidências de esplenomegalias em curso frequentemente assintomáticas.
Nesse contexto, relatamos o caso de uma paciente com a forma adulta da doença NP-C, enfatizando suas manifestações neuropsiquiátricas, a evolução clínica e a resposta farmacológica, tanto com terapia específica com medicamentos antipsicóticos, como com a terapia de redução de substrato através do uso de aminoaçúcar, que diminui a quantidade de gangliosídeos cerebrais, um dos elementos envolvidos na fisiopatologia da doença.
RELATO DE CASO
Paciente feminino, 18 anos, encaminhada para investigação de quadro neuropsiquiátrico, associado a quedas frequentes e ataxia.
Trata-se da primeira filha de casal jovem e não consanguíneo. Durante o pré-natal, a genitora foi diagnosticada com hipertensão gestacional, controlada com medicação. Realizou-se parto com 37 semanas e 4 dias, devido a sofrimento fetal agudo, com peso de nascimento de 2.100g, estatura de 42cm; APGAR 8/9. Além disso, apresentou icterícia nas primeiras horas de vida, sem necessidade de fototerapia e distensão abdominal, permanecendo internada por cinco dias.
Aos 2 meses de vida, evoluiu com piora do quadro abdominal, devido a hepatoesplenomegalia e déficit ponderal (Figura 2A). Ademais, apresentou descamação das nádegas, acolia fecal e colúria, permanecendo hospitalizada por 60 dias. Após esse período, sem sinais de melhora, fez-se indicação clínica de transplante hepático, porém genitores declinaram do procedimento. Paulatinamente, contudo, houve gradual melhora do quadro de hepatoesplenomegalia (Figura 2B), com normalização do quadro de colestase.
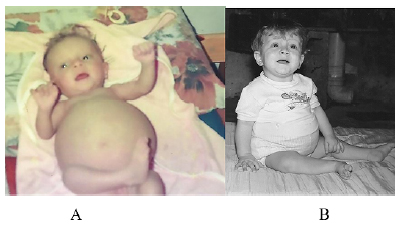
Figura 2. A) Paciente com importante hepatoesplenomegalia aos 2 meses de vida B) Melhora gradual da hepatoesplenomegalia e resolução espontânea da colestase aos 8 meses de vida
Fonte: os autores (2021)
Aos 2 anos de idade, no entanto, mantinha hepatoesplenomegalia, e foi realizada biópsia de fígado, cujo resultado foi inconclusivo, sugerindo tratar-se de "uma doença metabólica hereditária". No decorrer dos anos, a paciente evoluiu com remissão do quadro de visceromegalia e normalização da função hepática, sem apresentar outras comorbidades.
Marcos do neurodesenvolvimento da paciente foram compatíveis com a normalidade: sustento cefálico aos 3 meses, sentar sem apoio aos 6 meses, engatinhar aos 9 meses, fala com 12 meses e andar sem apoio aos 18 meses.
Aos 18 anos de idade, apresentou quadro de depressão, que evoluiu com sintomas psicóticos ("esquizofrenia-like"), e foi iniciado acompanhamento com psiquiatra. Seu quadro clínico tornou-se mais complexo, aos 22 anos, passando a cursar com alucinações auditivas e visuais, insônia, agressividade e sintomas persecutórios. Foram introduzidos Clonazepam e Risperidona para manejo dos sintomas psicóticos, sendo, posteriormente, suspensos devido a alterações hepáticas.
Progressivamente, passou a cursar com episódios de tropeços, quedas, sintomas de incoordenação motora e de postura anormal de mãos. Houve também declínio cognitivo, que foi considerado como secundário ao quadro psiquiátrico de base, mas que não afetava as atividades diárias da paciente, embora tenha passado também a ficar mais cada vez mais "esquecida" (sic), em particular com dificuldades para recordar-se de acontecimentos recentes.
Seu exame físico, à época, evidenciava: fáscies simétricas com presença de movimentos distônicos faciais; paresia ocular supranuclear vertical, leve disartria; força muscular grau 5 em membros superiores e inferiores; reflexos osteotendíneos vivos globalmente; dismetria; disdiacocinesia; presença de postura distônica em mãos; aumento de tônus em membros inferiores; reflexo cutâneo-plantar em flexão; sensibilidade profunda e superficial preservadas; marcha com base alargada.
Diante dos achados do exame físico e da história clínica, fez-se a suspeita de doença de Niemann-Pick tipo C, procedendo-se à realização da biópsia de pele para a cultura de fibroblastos, a fim de realizar o teste de Filipin; coleta de sangue para pesquisa molecular de mutações do gene NPC1 e NPC2. Teste do Filipin revelou-se positivo, com presença de intensa fluorescência perinuclear corroborando o diagnóstico de NP-C, que foi confirmado pelo sequenciamento genético, o qual evidenciou a presença de 2 mutações missense do gene NPC1: a mutação c.1114C>T p.R372W do éxon 8 e a mutação c.3104C>T p.A1035V do éxon 2. Confirmou-se, assim, o diagnóstico clínico e bioquímico da doença.
Após confirmação diagnóstica, iniciou uso de terapia de redução de substrato (TRS) com Miglustate, apresentando estabilização clínica dos sintomas neurológicos de ataxia cerebelar e distonia. Após introdução desse medicamento, sintomas psiquiátricos, em combinação com tratamento utilizando antipsicóticos, obtiveram melhor resposta terapêutica com remissão de sintomas de alucinação auditiva e comportamento persecutório, embora ainda tenha apresentado sintomas depressivos leves.
Durante todo esse processo, a paciente iniciou reabilitação motora, natação, fisioterapia, fisiatria, equoterapia, terapia ocupacional, musicoterapia e acompanhamento com psicólogo, nutricionista, fonoaudiólogo e psicopedagogo.
DISCUSSÃO
Niemann-Pick tipo C (NP-C; OMIM 257220) é uma doença hereditária autossômica recessiva que afeta o metabolismo lipídico, ocasionando o distúrbio de armazenamento lisossomal neurovisceral, devido ao defeito no trânsito celular ,que leva a um acúmulo de glicoesfingolipídios e colesterol em diversos órgãos.3,4
A NP-C é uma doença com grande variabilidade fenotípica; os sintomas neurológicos são bastante comuns, abrangendo desde uma apresentação clínica com manifestações neuropsiquiátricas de início juvenil a quadros psiquiátricos adultos semelhantes a esquizofrenia.11,12
Vale ressaltar que há um fenótipo da doença com alterações viscerais (como hepatoesplenomegalia) e colestase prolongada no primeiro ano de vida, sem ainda ocorrer manifestações neurológicas. Portanto, com base nessas características, deve-se considerar a NP-C como diagnóstico diferencial de pacientes com quadro visceral de organomegalia e/ou colestase prolongada nos primeiros meses de vida, sem causa definida. Embora sintomas neurológicos tendam a surgir no curso da doença, eles podem aparecer apenas décadas depois do quadro visceral (o qual pode também remitir ao longo do tempo), como foi o caso da paciente do relato.
A suspeita clínica de NP-C precisa ser corroborada pela confirmação bioquímica ou genético-molecular. A pesquisa de células espumosas (figura 3) em esfregaço de medula óssea é um exame historicamente utilizado para investigação de NP-C, embora seja pouco específico, visto que outras doenças lisossomais podem apresentar células espumosas ao exame de medula óssea.
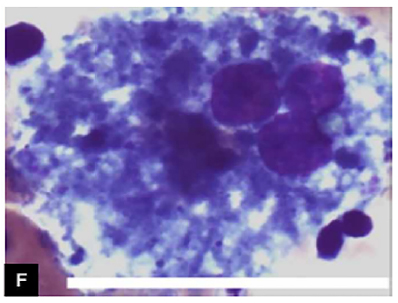
Figura 3. May- Coloração de Grümwald-Giemsa, ou células Niemann-Pick. Barra = 50um.
Fonte: Imagem adaptada.10
Durante muito tempo, o teste de Filipin foi considerado o padrão-ouro para diagnóstico de NP-C. Consistia em um teste citoquímico bastante laborioso, que requeria biópsia de pele com cultivo de fibroblastos.4 O filipin é um corante que marca colesterol não esterificado, apresentando dois tipos de padrão como visualizado na figura 3. Paciente do relato exibiu "padrão clássico positivo" com intensa fluorescência perinuclear (figura 4A), o que já confirmaria a suspeita clínica de NP-C. No entanto, cerca de 15% dos pacientes com NP-C podem apresentar o padrão "variante" (figura 4B), o que requer a confirmação com análise genético-molecular para definição diagnóstica.
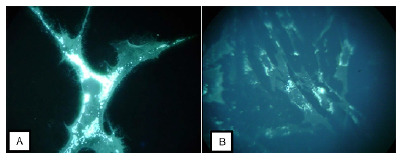
Figura 4. A- Padrão clássico de Filipin positivo; B- Padrão Variante de Filipin positivo.
Fonte: os autores (2019).
Em virtude do caráter laborioso do teste de Filipin e da presença não tão rara do padrão variante que exigia complementação diagnóstica, foram desenvolvidas outras ferramentas para o diagnóstico de NP-C, com o uso de biomarcadores (como a dosagem da "lysoesfingomielina"). Atualmente, usa-se o método de biomarcadores e análises genéticas como padrão ouro para diagnóstico de NP-C, seguindo-se um fluxograma de investigaçao9 (Figura 5).
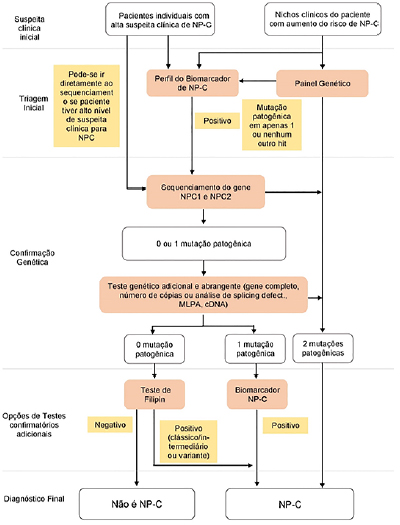
Figura 5. Fluxograma diagnóstico para NP-C
Fonte: imagem adaptada.9
No atual fluxograma de investigação de NP-C (figura 5), o teste de Filipin apenas é realizado em testes em que os estudos genéticos não foram conclusivos ou para validar mutações novas cujo significado clínico é incerto (e nas quais os biomarcadores não mostraram anormalidades).
Em relação ao quadro neuropsiquiátrico de NP-C, pode-se considerar que há uma ampla apresentação clínica, envolvendo desde sintomas pouco específicos, como deficiência intelectual, depressão, esquizofrenia, déficit de atenção, ataxia, distonia, hipotonia, mioclonias e epilepsia, até sintomas mais específicos, como cataplexia gelástica e paralisia do olhar conjugado supranuclear vertical (VSGP). Este último é um sintoma extremamente frequente entre os pacientes acometidos por essa doença na fase adulta.4,6-8 Na paciente do relato, não houve manifestações neurológicas prévias ao desenvolvimento do quadro psiquiátrico e de distúrbio de movimento a partir dos 18 anos de idade. A presença de VSGP ao seu exame físico, porém, foi uma pista importante para a suspeita de NP-C e deve ser um sinal clínico pesquisado em todo paciente com quadro neuropsiquiátrico progressivo sem causa estabelecida.
Em NP-C, não raro, a deterioração cognitiva pode ser facilmente confundida com demências neurodegenerativas ou distúrbios psiquiátricos, sendo de suma importância essa distinção.13-16 Pacientes adultos com NP-C podem manifestar - como sintoma inicial e principal - quadro de deterioração cognitiva progressiva mesmo na ausência de sintomas extrapiramidais ou de outros sintomas psiquiátricos. Embora a paciente descrita no relato tenha apresentado declínio cognitivo, o mesmo foi associado inicialmente como secundário ao quadro psiquiátrico, e não como parte da manifestação de sua doença de base. Sintomas demenciais, como por exemplo, de perda de memória recente, também são frequentes em pacientes com NP-C, em particular pacientes com "forma adulta" da doença.13
No caso relatado, a paciente exibiu sintomas viscerais pediátricos da doença, como hepatoesplenomegalia, icterícia e distensão abdominal, mas as manifestações cognitivas/neurodegenerativas se pronunciaram apenas na fase adulta, como "esquizofrenia-like" e VSGP.17 Assim, mesmo a paciente apresentando um quadro visceral inicial sugestivo de NP-C, foi diagnosticada tardiamente, provavelmente devido à normalização do quadro de colestase neonatal que havia manifestado na infância. Apenas quando surgiram as manifestações neuropsiquiátricas, a paciente foi encaminhada para nova investigação e houve a suspeita de NP-C.
Torna-se importante incluir na anamnese de pacientes adultos com quadros neuropsiquiátricos progressivos questionamentos quanto à presença de sintomas viscerais ou de colestase prolongada no primeiro ano de vida, pois são dados clínicos que podem ajudar a direcionar a investigação do paciente para NP-C.17
A única terapia específica aprovada para NP-C é o Miglustate. Esse medicamento atua como inibidor reversível e competitivo da glicosilceramida sintase, inibindo a síntese e acúmulo de glicoesfingolipídios cerebrais, que estabilizam ou retardam a progressão de danos neurológicos irreversíveis, comprovando assim a necessidade de implantação precoce do tratamento.18-22 No caso descrito, a paciente apresentou estabilização clínica dos sintomas neurológicos com o uso da medicação, mas é importante se considerar que, por se tratar de uma doença de caráter progressivo, melhor prognóstico se correlaciona com diagnóstico precoce, antes que sequelas neurológicas irreversíveis da doença estejam presentes.
Além disso, a paciente faz uso de outras terapias farmacológicas complementares ao Miglustate visando à melhora, principalmente, dos sintomas neuropsiquiátricos, como Risperidona e Clonazepam. Vale ressaltar a realização de terapêuticas não farmacológicas, como musicoterapia, equoterapia e psicoterapia, as quais também auxiliaram no quadro psiquiátrico.
Em suma, NP-C deve ser um diagnóstico diferencial importante a se considerar em pacientes com manifestações neuropsiquiátricas progressivas, em particular com histórico clínico prévio de colestase neonatal prolongada e visceromegalia. Por se tratar de uma enfermidade potencialmente tratável e dado seu caráter progressivo e devastador, o diagnóstico precoce torna-se fundamental, não só para melhor prognóstico clínico da paciente, mas também para permitir aconselhamento genético familiar e investigação de outros membros da família sob risco de serem afetados, mas ainda sem manifestação clínica aparente.
REFERÊNCIAS
1 Dougherty M, Lazar J, Klein JC, Diaz K, Gobillot T, Grunblatt E. et al. Genome sequencing in a case of Niemann-Pick type C. Mol Case Stud 2016; 2: a001222.
2 Amaral I do SA, Moia L de JMP, Coelho EFA, Medeiros ZL de, Montoril M de FP, Araujo MTF. Relatório de caso: doença de Niemann-Pick com manifestações de insuficiência hepática. Rev Pan-Amaz Saúde 2010; 1. doi:10.5123/S2176-62232010000300017.
3 Niemann-Pick C Disease Gene Mutations and Age-Related Neurodegenerative Disorders. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0082879 (accessed 21 May2019).
4 Kheder A, Scott C, Olpin S, Hadjivassiliou M. Niemann-Pick type C: a potentially treatable disorder? Pract Neurol 2013; 13: 382-385.
5 Koens LH, Kuiper A, Coenen MA, Elting JWJ, de Vries JJ, Engelen M et al. Ataxia, dystonia and myoclonus in adult patients with Niemann-Pick type C. Orphanet J Rare Dis 2016; 11: 121.
6 Piroth T, Boelmans K, Amtage F, Rijntjes M, Wierciochin A, Musacchio T et al. Adult-Onset Niemann-Pick Disease Type C: Rapid Treatment Initiation Advised but Early Diagnosis Remains Difficult. Front Neurol 2017; 8. doi:10.3389/fneur.2017.00108.
7 Nevsimalova S, Malinova V. Cataplexy and Sleep Disorders in Niemann-Pick Type C Disease. Curr Neurol Neurosci Rep 2014; 15: 522.
8 Evans WRH, Hendriksz CJ. Niemann-Pick type C disease - the tip of the iceberg? A review of neuropsychiatric presentation, diagnosis and treatment. BJPsych Bull 2017; 41: 109-114.
9 Patterson MC, Clayton P, Gissen P, Anheim M, Bauer P, Bonnot O et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: An update. Neurol Clin Pract 2017; : 1.
10 Lorenzoni PJ, Cardoso E, Crippa ACS, Lourenço CM, Souza FTS, Giugliani R et al.http://www.scielo.br/scielo.php?script=sci_abstract&pid=S0004-282X2014000300214&lng=en&nrm=iso&tlng=es. Arq Neuropsiquiatr 2014; 72: 214-218.
11 Sevin M, Lesca G, Baumann N, Millat G, Lyon-Caen O, Vanier MT et al. The adult form of Niemann-Pick disease type C. Brain 2006; 130: 120-133.
12 Imrie J, Heptinstall L, Knight S, Strong K. Observational cohort study of the natural history of Niemann-Pick disease type C in the UK: a 5-year update from the UK clinical database. BMC Neurol 2015; 15: 257.
13 Schicks J, Muller Vom Hagen J, Bauer P, Beck-Wodl S, Biskup S, KrägelohMann I, et al. Niemann-Pick type C is frequent in adult ataxia with cognitive decline and vertical gaze palsy. Neurology. 2013; 80:1169-70.
14 Bauer P, Balding DJ, Klunemann HH, Linden DEJ, Ory DS, Pineda M et al. Genetic screening for Niemann-Pick disease type C in adults with neurological and psychiatric symptoms: findings from the ZOOM study. Human Molecular Genetics 2013; 22: 4349-4356.
15 Maubert A, Hanon C, Sedel F. Psychiatric disorders in adult form of Niemann-Pick disease type C. L'Encephale. 2016; 42:208-13.
16 Bergeron D, Poulin S, Laforce R Jr. Cognition and anatomy of adult Niemann-Pick disease type C: insights for the Alzheimer field. Cogn Neuropsychol. 2017; 35:1-14.
17 Nadjar Y, Hütter-Moncada AL, Latour P, Ayrignac X, Kaphan E, Tranchant C et al. Adult Niemann-Pick disease type C in France: clinical phenotypes and long-term miglustat treatment effect. Orphanet J Rare Dis 2018; 13: 175.
18 Stein VM, Crooks A, Ding W, Prociuk M, O'Donnell P, Bryan C et al. Miglustat Improves Purkinje Cell Survival and Alters Microglial Phenotype in Feline Niemann-Pick Disease Type C. J Neuropathol Exp Neurol 2012; 71: 434-448.
19 Wraith JE, Vecchio D, Jacklin E, Abel L, Chadha-Boreham H, Luzy C et al. Miglustat in adult and juvenile patients with Niemann-Pick disease type C: Long-term data from a clinical trial. Molecular Genetics and Metabolism 2010; 99: 351-357.
20 Pineda M, Wraith JE, Mengel E, Sedel F, Hwu WL, Rohrbach M, et al. Miglustat in patients with Niemann-pick disease type C (NP-C): a multicenter observational retrospective cohort study. Mol Genet Metab 2009; 98:243-9.
21 Patterson MC, Vecchio D, Prady H, Abel L, Wraith JE. Miglustat for treatment of Niemann-Pick C disease: a randomised controlled study. The Lancet Neurology 2007; 6: 765-772.
22 Actelion. Miglustat (Zavesca) Summary of Product Characteristics, EMA. (EudraPharm) 2010. http://www.ema.europa.eu/ema/index.jsp?curl=pages/medicines/human/medicines/000435/human_med_001171.jsp&murl=menus/medicines/medicines.jsp&mid=WC0b0 1ac058001d125. Accessed 31 Aug 2018.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()