Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(4) - Dezembro 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Relato de caso: apresentação cliníca atípica da deficiência da esfingomielinase ácida em paciente pediátrico
Atypical clinical presentation of acid sphingomyelinase deficiency in a pediatric patient: a case report
Pedro Pagan1; Juliana Sanchez1; Beatriz Nascimento1; Felipe Balestra1; Fernando Colucci1; Zumira Aparecida Carneiro1; Laura Vagnini2; Charles Marques Lourenco1,2,3
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - RIBEIRAO PRETO - SP - Brasil
2. Centro Paulista de Diagnostico e Pesquisa, Genetica Clínica - RIBEIRAO PRETO - SP - Brasil
3. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
Endereço para correspondência:
Recebido em: 17/12/2020
Aprovado em: 24/05/2021
Instituição: Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - RIBEIRAO PRETO - SP - Brasil
Resumo
INTRODUÇÃO: A deficiência da enzima esfingomielinase ácida (ASMD), também conhecida como doença de Niemann-Pick A/B, é uma doença rara, causada por mutações recessivas no gene SMPD1 do cromossomo 11, acometendo tanto crianças como adultos. A ASMD provoca o acúmulo de esfingomielina em vários tecidos do corpo, principalmente no sistema reticuloendotelial, sendo comum a presença de hepatoesplenomegalia e envolvimento do parênquima pulmonar em indivíduos portadores de ASMD tipo B. O objetivo deste relato é apresentar o caso de um adolescente portador de NP-B com fenótipo mais atenuado da doença, enfatizando os desafios para o diagnóstico clínico e laboratorial de formas menos agressivas da doença.
RELATO DE CASO: Paciente masculino, três anos, foi encaminhado para avaliação de quadro de hepatoesplenomegalia e crises respiratórias frequentes. Apresentava aos exames de tomografia computadorizada do tórax sinais de acometimento pulmonar. Paciente foi encaminhado para serviço de genética, sendo investigado para doenças de armazenamento lisossômico com dosagem de diversas enzimas.
DISCUSSÃO: Associação de hepatoesplenomegalia com acometimento pulmonar são achados típicos que devem levar à suspeita de ASMD tipo B, permitindo a identificação mais rápida dessa enfermidade ainda na infância. Dessa forma, com o diagnóstico e tratamento precoces, o paciente com ASMD tipo B terá uma melhora considerável em sua qualidade de vida e uma redução importante no risco de complicações cardiovasculares futuras.
Palavras-chave: Doença de Niemann-Pick tipo B. Deficiência de esfingomielinase ácida lisossomial. Doença de armazenamento lisossômico.
Abstract
BACKGROUND: Acid sphingomyelinase deficiency enzyme (ASMD), also known as Niemann-Pick disease (NP) types A/B, is a rare condition caused by mutations in the chromosome 11, SMPD1 gene, affecting both children and adults. Due to the inherited deficiency in the activity of acid the sphingomyelinase enzyme, there is an accumulation of sphingomyelin in the cellular reticuloendothelial system of different tissues, culminating in hepatosplenomegaly and involvement of the pulmonary parenchyma, especially in individuals with NP-B. This report aimed to present the case of an adolescent with NP-B with a more attenuated phenotype of the disease, emphasizing the challenges for the clinical and laboratory diagnosis of less aggressive forms of the disease.
CASE REPORT: A 3-year-old male patient presented with hepatosplenomegaly and frequent respiratory crises, followed by recurrent lower airway infection. Image evaluation showed signs of pulmonary involvement. The patient was referred to a medical genetics' unit, where an investigation of lysosomal storage diseases was performed.
DISCUSSION: The association of hepatosplenomegaly with recurrent respiratory crises are indicative of NP-B. An early diagnosis leads to an improvement in the quality of life of the patients and a significant reduction in the risk of future cardiovascular complications. This case contributed to increasing disease awareness among clinical pediatricians and other healthcare professionals.
Keywords: Niemann-Pick type B disease. Lysossomal acid sphyngomylinase deficiency. Lysosomal storage disease.
INTRODUÇÃO
A deficiência da enzima esfingomielinase ácida (ASMD; OMIM #607616) também conhecida como doença de Niemann-Pick A/B, é uma doença rara, com prevalência de 0,4-1,0/100.000 no mundo, acometendo tanto crianças como adultos.1,2 O tipo A/B é causado pela deficiência de uma enzima lisossomal (esfingomielinase ácida), podendo apresentar sintomas neurológicos e visceromegalia.2,3 Já o tipo C consiste em uma disfunção do transporte de lipídios provocado pela mutação nos genes NPC1 e NPC2, que leva ao acúmulo de colesterol não esterificado e de esfingolipídios no fígado, baço e cérebro.2,3
A ASMD provoca o acúmulo de esfingomielina em vários tecidos do corpo (Figura 1), principalmente no sistema reticuloendotelial.4,5 Esse acúmulo de esfingolipídios faz com que as células da medula óssea tenham uma aparência "espumosa", sendo chamadas de células de Niemann-Pick.6,7
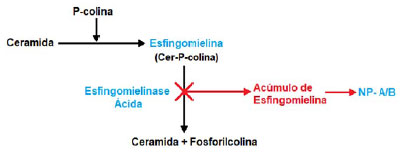
Figura 1. Via metabólica de degradação da esfingomielina.
Fonte: Os autores (2020).
Existem dois tipos principais de ASMD, ambos causados por mutações recessivas no gene SMPD1, no cromossomo 11.1,7,8 a recombinant human acid sphingomyelinase (rhASM O tipo A (NP-A) é caracterizado por um prognóstico mais grave devido à rápida degeneração do sistema nervoso central, além de acúmulo maciço de esfingolipídios no fígado e no baço, ocasionando hepatoesplenomegalia, com o óbito ocorrendo, na maioria dos casos, entre dois e três anos de idade.1a recombinant human acid sphingomyelinase (rhASM,7,8 Já o tipo B (NP-B) possui prognóstico menos grave, e apesar de não ser isento de complicações clínicas, normalmente não há o acometimento do sistema nervoso central, mas a presença de hepatoesplenomegalia e envolvimento do parênquima pulmonar é comum, sendo que indivíduos com a doença de NP-B possuem uma expectativa de vida maior em relação aqueles com NP-A.6,8,9
O propósito deste relato é apresentar o caso de um adolescente portador de NP-B com fenótipo mais atenuado da doença, enfatizando os desafios para o diagnóstico clínico e laboratorial de formas menos agressivas da doença e a importância do diagnóstico diferencial em indivíduos que apresentam esplenomegalia sem causa definida, principalmente nos pacientes pediátricos.
RELATO DE CASO
Paciente masculino, atualmente com 11 anos, adotado com dez semanas de vida, foi aos três anos foi encaminhado para avaliação de quadro de hepatoesplenomegalia. Nasceu de parto cesárea, a termo (37 semanas), comprimento de 47cm (percentil 25), peso de 3,150 kg (percentil 50), perímetro cefálico de 35cm (percentil 75), Apgar 9 no 5º minuto. Teve alta com 48 horas de vida. Não houve relato de intercorrências no período neonatal.
Apresentou atraso no desenvolvimento psicomotor, engatinhou com 11 meses, andou com um ano e nove meses e iniciou fala de frases com dois anos e cinco meses após iniciar fonoterapia. Atualmente, contudo, apresenta bom rendimento escolar e não há queixas quanto ao aprendizado.
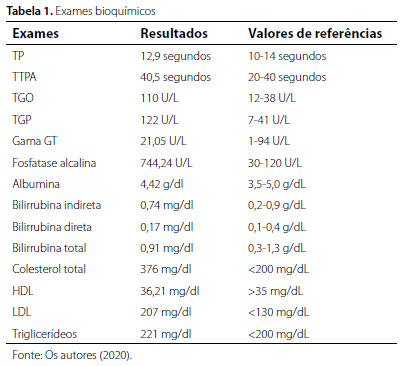
Aos três meses de idade, iniciou acompanhamento pediátrico em decorrência de "problemas respiratórios recorrentes e anemia crônica" (sic). Com um ano e cinco meses de idade, ao exame físico, observou-se presença de fígado a 6 cm do rebordo costal direito e baço a 2 cm do rebordo costal esquerdo. Em função desse achado, realizou-se ultrassonografia de abdome a qual confirmou hepatoesplenomegalia. Solicitaram-se exames bioquímicos para investigação de função hepática (Tabela 1) e, posteriormente, em exame de mielograma, observou-se presença de inúmeros macrófagos, histiócitos e várias células com conteúdo sugestivo de material gorduroso.
Como o quadro de hepatoesplenomegalia ainda não estava elucidado, aos três anos de idade, paciente foi submetido à biópsia do fígado, cujo resultado foi compatível com presença de tecido hepático fibroso com aglomerados de células volumosas e citoplasma espumoso, sugestivo de uma doença metabólica hereditária.
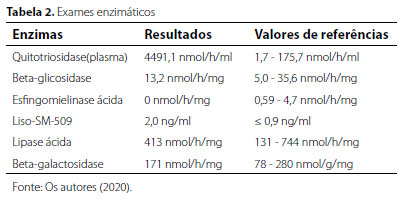
Paciente foi encaminhado para serviço de genética, sendo investigado para doenças de armazenamento lisossômico com dosagem de diversas enzimas, conforme Tabela 2, o qual foi diagnosticado ASMD tipo B. Estudo genético evidenciou presença de mutação do cromossomo 11, homozigótica no éxon 6 do gene SMPD1, c.1829_1831delGCC (p. Arg610del), confirmando diagnóstico bioquímico prévio. Por conseguinte, foi realizado acompanhamento médico, com tratamento sintomático do paciente.
Apresentou, no decurso de sua história clínica, diversas infecções de vias aéreas superiores (faringite e laringite com estridor), sempre acompanhadas de febre e intensa secreção, sendo indicada a retirada das tonsilas faríngeas aos cinco anos de idade. Possuía, ocasionalmente, episódios de sudorese, acompanhado de palidez e hipotermia, sem relação com perda de consciência ou outros fatores desencadeadores.
Após diagnóstico de NP-B, aos sete anos de idade, passou-se a realizar acompanhamento com exames de tomografia computadorizada do tórax; exames já evidenciavam sinais de acometimento pulmonar (Figura 2). Concomitantemente, foram realizadas espirometrias cujos resultados foram compatíveis com a normalidade e permanecem sem alteração.
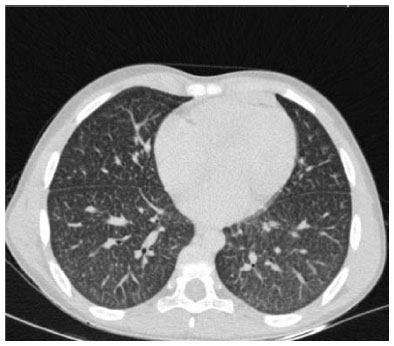
Figura 2. Tomografia computadorizada do pulmão: presença de opacidades reticulares e vidro fosco difusas em lobo médio e inferiores, com espessamento discreto nodular dos septos interlobulares Fonte: Os autores (2020).
Atualmente, com 11 anos e dois meses de idade, ainda apresenta quadros de infecção respiratória e de sudorese, porém com menor frequência e sem uso de medicações associado. Ao exame físico, peso 49,1kg (p90), estatura 1,55m (p90), aparelho respiratório sem alterações e abdômen flácido, com redução do quadro de hepatoesplenomegalia como característica atípica da história natural da doença.
DISCUSSÃO
A deficiência da esfingomielinase ácida (ASMD) é uma doença hereditária autossômica recessiva rara, sem predileção por gênero.10 ASMD tipo A é um distúrbio neurodegenerativo com hepatoesplenomegalia maciça e curso neurológico rapidamente progressivo, levando à morte por volta dos três anos de idade. ASMD tipo B, por sua vez, é um distúrbio não neuropático que causa hepatoesplenomegalia e comprometimento pulmonar.11 Os pacientes geralmente atingem a idade adulta, não havendo comprometimento do SNC. Ambos os tipos A e B são causados por mutações no gene SMPD1, ao contrário do tipo C, causado por mutações nos genes NPC1 e NPC2.11
O diagnóstico, antigamente, era realizado por meio de biópsia da medula óssea (BM), revelando acúmulo de macrófagos cheios de lipídios, conhecidos como histiócitos azul marinho.10 Mais modernamente, porém, vem se utilizando o teste enzimático em que há a demonstração de baixa atividade da esfingomielinase em ensaio enzimático de fibroblastos de pele cultivados ou, preferencialmente, leucócitos isolados a partir de amostra de sangue periférico, seguido do sequenciamento genético do SMPD1, que é o padrão-ouro.10
ASM ou esfingomielina fosfodiesterase-1 é codificada pelo gene SMPD1. Mais de 140 mutações no NPD foram relatadas (Human Gene Mutation Database: http://www.hgmd.org). Todos os pacientes diagnosticados com sintomas e baixa atividade ASM devem fazer uma análise genética, pois o prognóstico e até o manejo terapêutico podem diferir. Algumas formas podem ser mais graves, como insuficiência hepática e altos valores de colesterol, necessitando de intervenção médica.11
ASMD pode representar um desafio diagnóstico.12 O diagnóstico diferencial inclui doença de Wilson, deficiência de lipase ácida lisossomal, doença de Niemann-Pick tipo C e doença de Gaucher.12,13,14 Na deficiência de lípase acida lisossômica (LAL), o espectro clínico pode ser semelhante ao da ASMD, incluindo a alteração do perfil lipídico, mas o envolvimento pulmonar não é comumente relatado em pacientes com deficiência de LAL.12 No paciente relatado, foram realizados os exames laboratoriais das enzimas demonstradas na Tabela 2 para excluir os possíveis diagnósticos diferenciais, já que a ASMD não possui sinais e sintomas específicos para confirmar o diagnóstico clínico. No relato de caso apresentado, ASMD tipo B não foi a primeira hipótese diagnóstica, sendo direcionado devido aos resultados da quitotriosidase (plasma) elevada, um marcador inespecífico indicativo de doenças de depósito, e deficiência enzimática de esfingomielinase ácida. Por conseguinte, foi solicitado o estudo genético para a confirmação do diagnóstico.
O curso natural da doença hepática em ASMD tipo B, geralmente, não é progressivo, havendo estabilização do quadro clínico.13,14,15 No entanto, um subconjunto de pacientes é descrito na literatura, evoluindo para de insuficiência hepática.14,15 Foram relatados na literatura vários casos de pacientes adultos com ASMD que apresentavam cirrose hepática e hipertensão portal.15 A análise de pacientes que morreram de doença hepática ou tiveram transplante de fígado fornece evidências adicionais de que um subconjunto de pacientes com início dos sintomas na infância tem um risco aumentado de insuficiência hepática precoce, porém no caso apresentado, como paciente é muito jovem, ainda não se sabe se permanecerá estável ou poderá apresentar complicações hepáticas.15 Lipiński et al., contudo, apresentam que a morte por doença hepática em pacientes com ASMD tipo B em uma série de pacientes poloneses foi igualmente frequente em pacientes pediátricos e adultos, e todos, exceto um paciente adulto, morreram antes dos 45 anos.14 No caso apresentado, o paciente iniciou com hepatomegalia evidenciada ao exame físico e ultrassom de abdome. Entretanto, em sua evolução clínica, houve estabilização dos sintomas hepáticos com remissão da hepatoesplenomegalia, havendo pouco impacto no quadro clínico, sendo diferente dos fenótipos típicos da doença relatados na literatura.
Verificou-se que a maioria dos pacientes com ASMD apresenta doença pulmonar intersticial por métodos radiográficos.17 A fisiopatologia da doença pulmonar envolve o acúmulo de macrófagos carregados de lipídios nos espaços e septos alveolares, paredes brônquicas e pleura, resultando em restrição progressiva dos volumes pulmonares e no comprometimento das trocas gasosas.17 O envolvimento pulmonar faz parte da manifestação multissistêmica do armazenamento de esfingomielina.15,16 O paciente do relato apresentou quadros de crises respiratórias recorrentes e infecções de repetição em vias aéreas inferiores, típico no ASMD tipo B, diferente do quadro hepático atípico, ocorrendo permanência dos sinais e sintomas ao longo do desenvolvimento permanecendo como a principal queixa.
Os achados da tomografia de tórax mais comumente relatados na literatura são espessamento septal interlobular liso e linhas intralobulares, predominantemente nos lobos inferiores dos pulmões, e opacidades em vidro fosco focais ou difusas, também predominantemente nas zonas pulmonares inferiores.16,17 Na literatura, as alterações observadas na TC afetam mais comumente os lobos inferiores, bilateralmente.18 No paciente deste relato, os achados tomográficos pulmonares foram clássicos do quadro de ASMD tipo B, com presença de opacidades reticulares e vidro fosco difusas em lobo médio e inferiores, com espessamento discreto nodular dos septos interlobulares. O espessamento septal interlobular liso pode ser observado em muitas doenças venosas, linfáticas ou infiltrativas, sendo a mais comum o edema pulmonar.19 O conhecimento das diferentes causas desse padrão pode ser útil na prevenção de erros de diagnóstico.19 Além disso, embora as causas desse padrão sejam muitas vezes indistinguíveis pela avaliação radiológica, diferenças na distribuição das lesões nos pulmões, achados radiológicos associados, histórico do paciente e apresentação clínica podem ser úteis para sugerir o diagnóstico apropriado.18
Os principais diagnósticos diferenciais desse tipo de padrão pulmonar, conhecido como padrão de pavimentação em mosaico ("crazy pattern"), são doenças adquiridas de causas aguda ou crônica.20 As principais condições agudas que causam padrão de pavimentação em mosaico são: edema pulmonar; infecções agudas (pneumocistose, leptospirose, pneumonia, H1N1) e hemorragia alveolar difusa.20 As principais condições crônicas que geram um padrão de pavimentação em mosaico são: adenocarcinoma invasivo, pneumonia lipoide, carcinomatose linfangítica e proteinose alveolar.20 As alterações histopatológicas correspondem à presença de macrófagos espumosos nos septos interlobulares e a algum grau de fibrose intersticial, resultando em uma aparência de espessamento septal.16 As mesmas células que envolvem os alvéolos estão associadas a opacidades em vidro fosco.16 No caso do paciente do relato, por apresentar um padrão de pavimentação em mosaico, foi realizada investigação do acometimento pulmonar, afastando causas infecciosas e, posteriormente, diante da cronicidade do quadro e da associação com outros sintomas, como hepatoesplenomegalia, direcionou-se a hipótese diagnóstica para doenças genéticas de depósito que pudessem cursar com a combinação desses achados, como, a doença de Gaucher, ASMD tipo A, ASMD tipo B e ASMD tipo C.
Até o momento, não há uma terapia curativa para ASMD tipo B. O uso de estatinas pode ser indicado para manejo da dislipidemia em alguns pacientes, embora não existam estudos clínicos abordando o uso dessa medicação na população pediátrica.1 Recentemente, porém, desenvolveu-sevuma nova terapia de reposição enzimática (TRE) para ASMD tipo B. Tal abordagem, que envolve a substituição da enzima deficiente, a esfingomielinase ácida, por uma forma recombinante geneticamente modificada (olipudase alfa), demonstrou resultados promissores na redução dose-dependente dos níveis tissulares de esfingomielina em doses de até 3.0 mg/kg com efeitos adversos de leves a moderados. Entretanto, a medicação está em fase de estudo clínico, que ainda não foi concluído, e não pode ser adquirida para uso.1,21
Ademais, a olipudase alfa demonstrou boa tolerância em um tratamento de 30 meses, associada a importante melhora na qualidade de vida e nos parâmetros clínicos dos pacientes avaliados, sem a ocorrência de reações de sensibilidade ou reações imunológicas contra o medicamento.21,22 Além disso, foi descrita uma redução estatisticamente importante no volume hepático (31,2%) e esplênico (39,3%), comparável às respostas observadas em estudos que avaliaram a aplicação de TRE em outros distúrbios de armazenamento lisossomal, assim com um aumento de 35% na capacidade funcional pulmonar dos pacientes.21,23
O seguimento do paciente com o diagnóstico de NP-B envolve uma equipe multidisciplinar focada nas principais manifestações da doença e seu impacto sobre a saúde do indivíduo, conforme observado na figura 3.
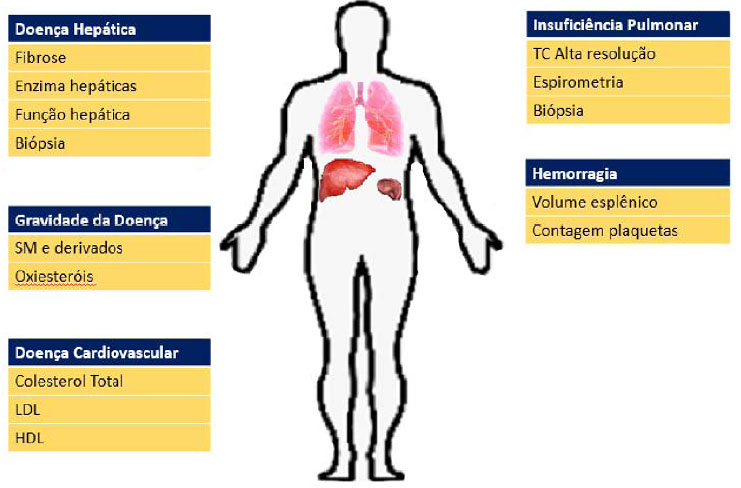
Figura 3. Seguimento dos acometimentos de NP-B. Fonte: Os autores (2020).
Em suma, pacientes pediátricos e adultos apresentando quadro de hepatoesplenomegalia associado ao padrão de infiltração pulmonar de pavimentação em mosaico e/ou opacidade em vidro fosco devem ter a ASMD tipo B como possível diagnóstico, considerando o recente progresso no desenvolvimento de uma TRE eficiente para este distúrbio de armazenamento lisossomal. Dessa forma, com o diagnóstico e tratamento precoce, o paciente com ASMD tipo B terá uma melhora considerável em sua qualidade de vida e uma redução importante no risco de complicações cardiovasculares futuras.
REFERÊNCIAS
1. Wasserstein MP, Jones SA, Soran H, Diaz GA, Lippa N, Thurberg BL, et al. Successful within-patient dose escalation of olipudase alfa in acid sphingomyelinase deficiency. Mol Genet Metab [Internet]. 2015;116(1-2):88-97
2. Geberhiwot T, Moro A, Dardis A, Ramaswami U, Sirrs S, Marfa MP, et al. Consensus clinical management guidelines for Niemann-Pick disease type C. Orphanet J Rare Dis. 2018;13(1):1-19
3. Cerón-Rodríguez M, Vázquez-Martínez ER, García-Delgado C, Ortega-Vázquez A, Valencia-Mayoral P, Ramírez-Devars L, et al. Niemann-Pick disease A or B in four pediatric patients and SMPD1 mutation carrier frequency in the Mexican population. Ann Hepatol. 2019;18(4):613-9.
4. Patiño-Escobar B, Solano MH, Zarabanda L, Casas CP, Castro C. Niemann-Pick Disease: An Approach for Diagnosis in Adulthood. Cureus. 2019 ;11(5): 2-7.
5. Chuang WL, Pacheco J, Cooper S, McGovern MM, Cox GF, Keutzer J, et al. Lyso-sphingomyelin is elevated in dried blood spots of Niemann-Pick B patients. Mol Genet Metab [Internet]. 2014;111(2):209-11.
6. Minai OA, Sullivan EJ, Stoller JK. Pulmonary involvement in Niemann-Pick disease: Case report and literature review. Respir Med. 2000;94(12):1241-51.
7. Schuchman EH, Disease G. Best Practice & Research Clinical Endocrinology & Metabolism Types A and B Niemann-Pick disease. Best Pract Res Clin Endocrinol Metab [Internet]. 2015;29(2):237-47.
8. Simões RG, Maia H. Niemann-Pick type B in adulthood. BMJ Case Rep. 2015;2015:2014-6.
9. Wasserstein M, Godbold J, McGovern MM. Skeletal manifestations in pediatric and adult patients with Niemann Pick disease type B. J Inherit Metab Dis. 2013; 36(1):123-7.
10. Irun P, Mallen M, Rodriguez-Sureda V, Alvarez-Sala LA, Arslan N, Bermejo N, Guerrero C, Perez de Soto I, Villallon L, Dominguez C, Giraldo P, Pocovi M. Identification and characterization of six novel SMPD1 mutations causing Niemann-pick types a/B disease. Clin Genet. 2013; 84:356-61.
11. Schuchman EH, Desnick RJ. Niemann Pick Disease type A and B: acid sphyngomyelinases deficiencies. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The metabolic and molecular basis of inherited disease. 8th ed. New York: McGraw-Hill; 2001; 3589-610.
12. Reiner Z, Guardamagna O, Nair D, Soran H, Hovingh K, Bertolini S, Jones S, Ćorić M, Calandra S, Hamilton J, Eagleton T, Ros E. Lysosomal acid lipase deficiency. An under-recognized cause of dyslipidaemia and liver dysfunction. Atherosclerosis. 2014; 235:21-30
13. Eskes ECB, Sjouke B, Vaz FM, Goorden SMI, van Kuilenburg ABP, Aerts JMFG, Hollak CEM. Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Mol Genet Metab. 2020;130(1):16-26.
14. Lipiński P, Kuchar L, Zakharova EY, Baydakova GV, Ługowska A, Tylki-Szymańska A. Chronic visceral acid sphingomyelinase deficiency (Niemann-Pick disease type B) in 16 Polish patients: long-term follow-up. Orphanet J Rare Dis. 2019;14(55); 1-9.
15. Cassiman D, Packman S, Bembi B, Turkia HB, Al-Sayed M, Schiff M, Imrie J, Mabe P, Takahashi T, Mengel KE, Giugliani R, Cox GF. Cause of death in patients with chronic visceral and chronic neurovisceral acid sphingomyelinase deficiency (Niemann-Pick disease type B and B variant): Literature review and report of new cases. Mol Genet Metab. 2016;118(3):206-13.
16. Baldi BG, Santana AN, Takagaki TY, Fujita C, Kairalla RA, Carvalho CR. Lung cyst: an unusual manifestation of Niemann-Pick disease. Respirology. 2009;14(1):134-6.
17. Mendelson DS, Wasserstein MP, Desnick RJ, Glass R, Simpson W, Skloot G, et al. Type B Niemann-Pick disease: findings at chest radiography, thin-section CT, and pulmonary function testing. Radiology. 2006;238(1):339-45.
18. Simpson WL Jr, Mendelson D, Wasserstein MP, McGovern MM. Imaging manifestations of Niemann-Pick disease type B. AJR Am J Roentgenol. 2010;194(1): W12-9
19. Guillemot N, Troadec C, de Villemeur TB, Clément A, Fauroux B. Lung disease in Niemann-Pick disease. Pediatr Pulmonol. 2007;42(12):1207-14.
20. Rodrigues R, Marchiori E, Müller NL. Niemann-Pick disease: high-resolution CT findings in two siblings. J Comput Assist Tomogr. 2004;28(1):52-4.
21. Wasserstein MP, Diaz GA, Lachmann RH, Jouvin M-H, Nandy I, Ji AJ, et al. Olipudase alfa for treatment of acid sphingom yelinase deficiency (ASMD): safety and efficacy in adults treated for 30 months. J of Inherited Metabolic Disease. 2018;41(5):829-38.
22. Diaz G, Bembi B, Giugliani R, Guffon N, Jones S, Cowan L, et al. Preliminary data from first clinical trial of enzyme replacement therapy with olipudase alfa in pediatric patients with chronic visceral and neurovisceral acid sphingomyelinase deficiency. Molecular Genetics and Metabolism. 2020;129(2):S48-9.
23. Thurberg BL, Diaz GA, Lachmann RH, et al. Long-term efficacy of olipudase alfa in adults with acid sphingomyelinase deficiency (ASMD): Further clearance of hepatic sphingomyelin is associated with additional improvements in pro- and anti-atherogenic lipid profiles after 42months of treatment, Molecular Genetics and Metabolism .2020; 1-29
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()