Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(4) - Dezembro 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Atrofia muscular espinhal com fraturas congênitas 2: Relato do primeiro paciente latino-americano e revisão da literatura
Spinal Muscular Atrophy With Congenital Bone Fractures 2: A Case Report Of The First Latin American Patient
Mattheus Lucca Amaral1, Mariana Dentelo Del-Campo1; Lucas Ribeiro Rodrigues1; Tainá Regina Damaceno Silveira2; Zumira Aparecida Carneiro1; Laura Vagnini3; Jordana Resende4; Regina Albuquerque4; Debora de Cassia Tomaz4; Marcela Almeida1; Fernanda Veiga Gomes5; Ana Paula Andrade Hamad6; Charles Marques Lourenco1,3,4
DOI:10.31365/issn.2595-1769.v21i4p218-222
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - RIBEIRÃO PRETO - SP - Brasil
2. Centogene AG, Genética Molecular - Rostock - Mecklemburgo-Pomerânia Ocidental - Alemanha
3. Centro Paulista de Diagnóstico e Pesquisa, Genética Clínica - RIBEIRÃO PRETO - SP - Brasil
4. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
5. Instituto Nacional de Saúde da Mulher, Criança e do Adolescente Fernandes Figueira (IFF) - Fiocruz, Neuropediatria - Rio de Janeiro - RJ - Brasil
6. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - RIBEIRÃO PRETO - SP - Brasil
Endereço para correspondência:
Recebido em: 19/11/2020
Aprovado em: 19/04/2021
Instituição: Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - RIBEIRÃO PRETO - SP - Brasil
Resumo
INTRODUÇÃO: Em 2016, dois grupos de autores publicaram os primeiros relatos acerca de recém-nascidos acometidos por uma nova manifestação de atrofia muscular espinhal com presença de fraturas ósseas congênitas. Essa condição é raríssima em neonatos e se apresenta como consequência de mutações autossômicas recessivas no gene ASCC1. Seu quadro clínico envolve hipocinesia/acinesia fetal, degeneração neuromotora, hipotonia, fraturas congênitas, contraturas articulares e curta expectativa de vida pós-parto, na maioria dos casos. Neste relato, apresenta-se o primeiro caso de atrofia muscular espinhal com fraturas congênitas (SMABF2) da América Latina.
RELATO DE CASO: Paciente, pré-termo de 33 semanas, masculino, filho de pais não consanguíneos, apresentou hipocinesia fetal, artrogripose de mãos e pés, hipotonia, afilamento de arcos costais e esforço respiratório no primeiro minuto de vida, dependente de ventilação mecânica e diagnóstico de atrofia muscular espinhal com fraturas congênitas (SMABF2).
DISCUSSÃO: Até o presente momento, existem 11 casos de SMABF2 descritos, e todos os pacientes apresentaram depressão respiratória neonatal, fraturas congênitas e artrogripose. Destaca-se a variedade da apresentação clínica dos pacientes previamente relatados, visto que a hipotonia neonatal e atrofia muscular não se manifestaram em todos os recém-nascidos. O paciente deste relato faz uso de ventilação mecânica constantemente, bem como se alimenta por sonda nasogástrica. Apesar de restrito ao leito desde o nascimento, vem apresentando expectativa de vida mais longa em comparação aos casos já publicados na literatura, em que a maioria faleceu logo após o nascimento ou com alguns dias de vida, estando com 1 ano e 4 meses de vida
Palavras-chave: Atrofias Musculares Espinais da Infância. Artrogripose. Fraturas do Fêmur. Sequenciamento Completo do Exoma
Abstract
BACKGROUND: Spinal muscular atrophy with congenital bone fractures (SMABF2, OMIM #616867) was firstly described in 2016 by two independent groups. The cases reported the medical journey of newborns affected by a new manifestation of spinal muscular atrophy with the presence of congenital bone fractures. This exceedingly rare condition is induced by autosomal recessive mutations in the ASCC1 gene, leading to fetal hypokinesia/akinesia, neuromotor degeneration, hypotonia, congenital fractures, joint contractures followed by a short postpartum life expectancy in most cases. Here, we report the first case of SMABF2 in Latin America.
CASE REPORT: A male, 33-weeks-old preterm patient, son of non-consanguineous parents, presented with fetal hypokinesia, arthrogryposis of hands and feet, hypotonia, thin ribs and respiratory effort in the first minute of life, depending on mechanical ventilation as well as nasogastric tube. The patient was then diagnosed with SMABF2.
DISCUSSION: To the best of our knowledge, there are eleven cases of SMABF2 in the literature. All patients had neonatal respiratory depression, congenital fractures and arthrogryposis. Besides these clinical manifestations, the patient presented neonatal hypotonia and muscle atrophy, which were not verified in all the previous case reports. Although restricted to bed since birth, life expectancy of the patient (1.3y) is longer than the previously reported (death shortly after birth or within a few days of life).
Keywords: Spinal Muscular Atrophies of Childhood. Arthrogryposis. Fractures, Bone. Whole Exome Sequencing
INTRODUÇÃO
As atrofias musculares são um dos principais grupos de doenças neuromotoras, no qual se encontram inseridas as atrofias musculares espinhais (AMEs). O quadro clínico desse grupo de doenças é caracterizado por fraqueza muscular progressiva, culminando em ausência de movimento, malformações ósseas e insuficiência respiratória. Há múltiplos fenótipos de AMEs, sendo sua classificação baseada no tempo de aparecimento da clínica e sua gravidade. As AMEs são, em 96% dos casos, causadas por mutações no gene SMN1, geralmente decorrente de deleção homozigótica nos éxons 7 e/ou 8. Como o gene SMN (Survival Motor Neuron) localiza-se no cromossomo 5q, usualmente denomina-se essa forma mais comum de AME de AME 5q13, para diferenciar das outras formas mais raras de amiotrofia espinhal causadas por mutações em outros genes.1-2
As AMEs que cursam com fraturas congênitas e artrogripose são um grupo mais raro de amiotrofias espinhais cuja etiologia genética vem sendo elucidada recentemente. Tanto mutações no gene SMN1 quanto em genes descritos nos últimos anos, como TRIP4 e ASCC1 (localizados respectivamente nos cromossomos 15q e 10q), estão implicados nessa forma de apresentação.
Do ponto de vista clínico, entre as formas de atrofia muscular espinhal com contraturas, a forma grave de AME por mutações no SMN1 chamada de AME 5q tipo 0 foi uma das primeiras descritas. Pacientes com essa forma de AME 5q possuem manifestações intrauterinas (com hipocinesia fetal, contraturas) e, ao nascimento, cursam com grave hipotonia e dificuldade respiratória, necessitando de suporte ventilatório ainda nas primeiras horas de vida.3-4
Entre as formas mais raras de AME "não 5q" com contraturas, destaca-se a atrofia muscular espinhal com fraturas congênitas (SMABF2, cromossomo 10q22) (OMIM #616867), causada por mutações autossômicas recessivas no gene ASCC1, com frequência de aproximadamente 10 entre 141 mil indivíduos da população mundial,5 cujo quadro clínico consiste de um conjunto de sinais e sintomas no qual se destaca a acinesia e/ou hipocinesia fetal como achado mais precocemente observado. Além disso, podem estar presentes: degeneração neuromotora, desenvolvimento anormal do esqueleto/tecido conjuntivo, hipotonia, fraturas ósseas congênitas, oligoâmnio e restrição do crescimento intraútero.1,5-6 Seu acometimento neurológico engloba a perda dos neurônios motores inferiores, do núcleo do tronco cerebral com desenvolvimento de artrogripose, sendo mais comumente presente a amioplasia (artrogripose clássica).7
Hipocinesia ou acinesia fetal acarreta o desenvolvimento de contraturas articulares. Na presença de duas ou mais contraturas, tem-se a definição de artrogripose, sendo o termo "artrogripose múltipla congênita" comumente utilizado em casos de múltiplas contraturas articulares.5,7-8 Casos mais graves da doença incluem manifestações como ausência de movimento espontâneo e insuficiência respiratória devido à incapacidade da musculatura respiratória.
Na patogênese da amiotrofia espinhal com fraturas congênitas tipo 2, há fortes evidências de que mutações no gene ASCC1 (activator signal cointegrator complex) (OMIM *614215) afetando sua interação com outros genes do complexo ASC-1, como o TRIP4 (OMIM *604501), são as responsáveis pelo desenvolvimento das alterações do neurônio motor presentes na doença.1-9 O ASC-1 (Figura 1) é um complexo de cointegração do processo de transcrição que é composto por quatro subunidades: TRIP4, ASCC1, ASCC2 e ASCC3. O ASC-1 parece agir nos eventos de maquinaria do RNA e, fundamentalmente, também na coativação do processo de transcrição gênica.9 Embora ainda não se tenha elucidado exatamente como esse processo leva à alteração neuronal, já há outros exemplos de doenças neuromusculares hereditárias causadas por mecanismos envolvendo o processo de transcrição/tradução genéticos e modulação do RNA.9
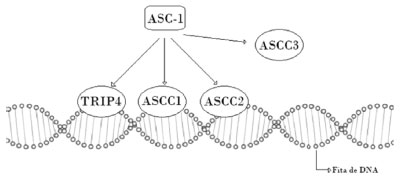
Figura 1. O complexo de cointegração transcricional ASC-1 é composto por quatro subunidades. TRIP4 (atua ligando os fatores de transcrição), ASCC1(participa religando a um domínio do RNA), ASCC2 (não muito conhecido), ASCC3 (RNA helicase). Fonte: os autores (2020).
Este artigo busca apresentar caso do primeiro paciente latino-americano portador de mutação autossômica recessiva em homozigose do gene ASCC1 e seu fenótipo clínico em comparação com os outros casos descritos na literatura.
RELATO DE CASO
Paciente do sexo masculino, caucasiano, filho de pais não consanguíneos, foi encaminhado para investigação de quadro de atrofia muscular espinhal com fraturas congênitas. Não há casos semelhantes na família, paciente possui dois irmãos hígidos (Figura 2A).
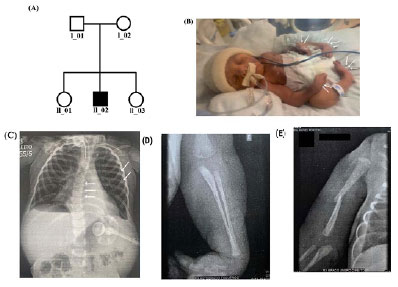
Figura 2. (A) Heredograma familiar (B) Setas apontam malformações bilaterais em região de metacarpos das mãos, bem como nos pés, que apresentam padrão de supinação. (C) Radiografia de tórax mostrando afinamento das costelas e escoliose em coluna torácica. (D) Radiografia AP de antebraço esquerdo (E) Radiografia AP de braço, úmero direito apresentando fratura de diáfise oblíqua com desvio medial. Fonte: os autores (2020)
Paciente nasceu de parto cesáreo em virtude de oligoâmnio progressivo, pré-termo (34 semanas), após uma gestação sem outras intercorrências, além do diagnóstico intrauterino de pés tortos congênitos e hipocinesia fetal. Genitora referiu pouca movimentação fetal durante a gestação. Criança nasceu com 1.790 gramas (<p10), 40,5cm de estatura (<p10) perímetro cefálico de 30,5cm (p25<p<p50), Apgar no 1° minuto de quatro, e no 5° minuto, de sete.
No parto, paciente apresentou síndrome a esclarecer composta por pés tortos bilateralmente, deformidade de metacarpos, artrogripose de mãos e pés, hipotonia acentuada, pescoço encurtado (Figura 2B), excesso de tecido adiposo em região torácica e tocotraumatismo com hematoma em hemitórax esquerdo posteriormente. Também foram constatados hipertelorismo mamário e criptorquidia bilateral, e afilamento de arcos costais (Figura 2C). Paciente evoluiu com depressão respiratória moderada no primeiro minuto e leve no quinto. Encontra-se em uso de ventilação mecânica (VM) desde o nascimento, inicialmente em VM não-invasiva evoluindo, após três meses, para traqueostomia. Posteriormente, inicia-se treino preparatório para BIPAP (BI-level positive airway pressure) associado à VM, associação que vem mantendo até o momento. Após 10 dias do parto, apresentou quilótorax, sendo realizada drenagem torácica em hemitórax direito em três momentos diferentes e, com um mês de vida, realizou-se drenagem do hemitórax esquerdo por quilotórax por três semanas.
Paciente recebeu alta após quatro meses em UTI neonatal, dependente de ventilação mecânica e BIPAP e sob dieta por sonda nasogástrica. Além da depressão respiratória, já exibia sinais evidentes de hipotonia muscular generalizada, acompanhada por fratura de úmero direito e contraturas articulares em mãos e pés, caracterizando quadro de artrogripose múltipla.
Após triagem e exclusão de mutações no gene SMN e de erros inatos do metabolismo, realizou-se sequenciamento completo de exoma para elucidação diagnóstica, sendo identificadas duas mutações frameshift em homozigose no gene ASCC1: c.157_158insG p.Glu53Gly fs*, confirmando o diagnóstico de SMABF2. Cada uma delas foi herdada de um dos genitores que, portanto, eram portadores heterozigotos.
Atualmente, paciente com idade de um ano e dois meses não apresentou nenhum dos marcos de desenvolvimento neuropsicomotor, sem intercorrências desde a alta hospitalar, em homecare contínuo. Ao exame físico, paciente apresenta: peso 7 kg (<p3), estatura 78cm (p50), perímetro cefálico 43cm (<p3), ausência de dismorfismos faciais significativos; fácies simétrico; motricidade ocular preservada, presença de fasciculações em língua; elevação simétrica de palato, hipotonia global; presença de contraturas em membros inferiores; reflexos osteotendíneos abolidos em membros superiores e inferiores; força muscular grau zero tanto em membros superiores quanto inferiores; sensibilidade preservada. Nível de atenção dentro da normalidade, atento ao ambiente externo e com boa interação visual com o examinador.
DISCUSSÃO
A SMABF2 congênita é uma doença neuromuscular autossômica recessiva caracterizada por hipotonia muscular, hipocinesia fetal, contraturas articulares múltiplas e fraturas majoritariamente de ossos longos. Recém-nascidos afetados comumente têm dificuldade para se alimentar e para respirar, e a insuficiência respiratória progressiva é fator determinante na letalidade da doença, sendo que a maioria dos recém-nascidos falece nos primeiros dias ou meses de vida.2-9, 12-13
O gene ASCC1 codifica a subunidade do complexo do sinal do cointegrador 1 de ativação (ASC-1). O ASC-1 é um complexo de cointegração do processo de transcrição, e a disrupção na expressão desse gene está associada ao desenvolvimento de outras patologias, como esôfago de Barret, adenocarcinoma esofágico e artrite reumatoide,9 também associado à patogênese da esclerose lateral amiotrófica multifatorial.11 Recentemente, mutações frameshift em homozigoze do gene ASCC1 foram relacionadas com quadros graves de artrogripose congênita associada com fraturas ósseas e de evolução letal. Mutações em outro gene, TRIP4 (OMIM 604501) no cromossomo 15q22, também foram identificadas como causa de uma rara forma de doença do neurônio motor inferior com fenótipo similar ao da forma grave neonatal da amiotrofia espinhal proximal, conhecida como SMABF1 (do inglês Spinal Muscular Atrophy with Congenital Bone Fractures type I).9 Na SMABF1, há duas mutações descritas que envolvem a troca de um nucleotídeo, c.760 C>T (p.Arg254*) e c.832C>T (p.Arg278), alterando a base nitrogenada.9 O acometimento de ambas as síndromes é semelhante, não havendo evidências na literatura atual que possam estabelecer a diferenciação clínica entre elas.
Mutações no gene ASCC1 passaram a constituir a SMABF2, para diferenciar dos casos por mutação no gene TRIP4. As primeiras descrições dessa nova forma de atrofia muscular espinhal com artrogripose e fraturas ósseas foram em 2016, a partir de dois casos relatados por Knierim et al.;5 posteriormente, Oliveira et al.6 descreveram dois casos de artrogripose com fraturas ósseas na mesma família, sendo um natimorto.
Em 2018, Bohm et al.5 apresentaram mais seis pacientes afetados por SMABF2 em três famílias distintas. Duas dessas famílias eram consanguíneas, apresentando mutações em homozigose, e a terceira apresentava duas variantes em heterozigose composta, a mutação nonsense (c.466C>T /p.Arg156*) e uma recorrente mutação frameshift c.157dupG (p.Glu53fs19*) (Tabela 1).
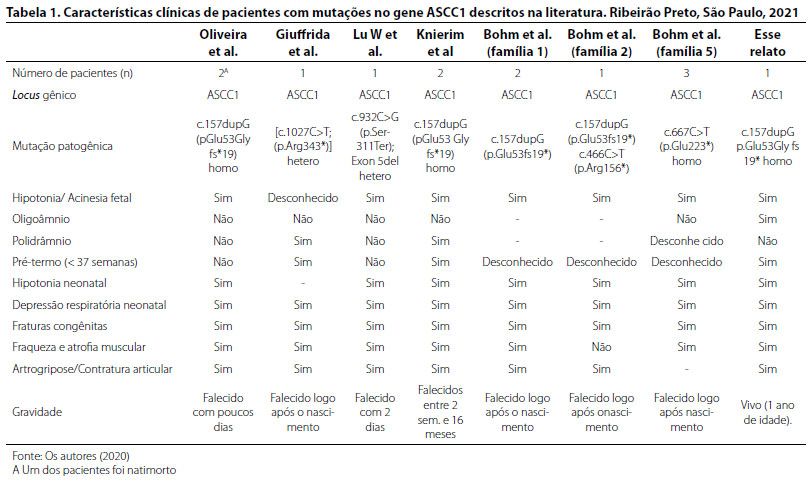
Há cinco mutações distintas descritas até o momento no gene ASCC1 ocasionando o fenótipo de SMABF2. A mutação identificada no paciente desse relato já fora previamente descrita na Tunísia por Bohm et al.5; em Portugal, por Oliveira et al.;6 e na Turquia, por Knierim et al.9
Todos os casos dessa doença descritos até o momento através de pesquisa em diferentes bases de dados de artigos médicos (PubMed, Scielo, Scopus, Web of Science, Lilacs, DOAJ, Google Scholar) foram resumidos na tabela 1. Todos os afetados apresentam artrogripose, fraturas congênitas, fraqueza, atrofia muscular e depressão respiratória neonatal. Dos 11 pacientes, hipotonia / acinesia fetal também foi identificada em dez pacientes; assim, essa condição também é descrita como atributo da atrofia muscular com múltiplas fraturas.1,5-10,12-13. Não é incomum a ocorrência de prematuridade nos casos de SMABF2: Giuffrida et al.12 e Knierim et al.,9 assim como o caso aqui relatado, descrevem o nascimento pré-termo em três crianças, enquanto Bohm et al.5 não fazem menção alguma à idade gestacional no momento do parto. Quanto à origem (nacionalidade) dos casos, foram descritos pacientes oriundos de diferentes países (inclusive Portugal e Espanha) e pertencentes a distintos grupos étnicos; não havia, entretanto, descrição de casos latino-americanos até o momento.
O paciente deste relato apresentou ainda hipotonia neonatal, múltiplas fraturas congênitas, atrofia muscular e artrogripose múltipla. Além disso, exibiu depressão respiratória ao nascer, que se perpetua até os dias hoje, fazendo uso de ventilação mecânica constantemente, bem como se alimenta por sonda nasogástrica. Esse paciente, apesar de restrito ao leito desde o nascimento, vem apresentando uma expectativa de vida mais longa em comparação aos casos publicados previamente, inclusive em relação a pacientes portadores da mesma mutação frameshift c.157dupG.
A apresentação clínica de SMABF2 possui vários pontos de sobreposição com outras formas de AME com contraturas ao nascimento, em particular com AME 5q tipo 0 como, por exemplo, hipocinesia fetal, artrogripose, dificuldade respiratória marcante nas primeiras horas de vida e grave hipotonia, o que fez com que mutações no gene SMN1 fossem inicialmente investigadas como causa do quadro clínico do caso relatado.
Há, no entanto, diferenças entre essas duas enfermidades, visto que, na AME 5q tipo 0, predominam achados de contraturas, edema periférico e dismorfias faciais (micrognatia, palato ogival), sendo menos frequentes restrição de crescimento intrauterino, fraturas e estreitamento torácico (achados que, por sua vez, são descritos com maior frequência em casos de SMABF2). Outra diferença notável na apresentação clínica entre as duas reside no fato de pacientes com AME 5q tipo 0 apresentarem maior incidência de cardiopatia congênita e de necrose digital (secundária à disautonomia), achados não descritos em SMABF2.
Em conclusão, relatamos aqui o primeiro caso de SMABF2 na América Latina, destacando o desenvolvimento de um quadro clínico clássico da doença, apresentando, contudo, um prognóstico mais favorável. Assim, todos os pacientes com atrofia muscular com fraturas congênitas de ossos longos devem ser avaliados inicialmente para amiotrofia espinhal por mutações no cromossomo 5q. Entretanto, caso essa possibilidade seja descartada, deve-se investigar outras síndromes neuromusculares, como a SMABF1 e 2, que são, respectivamente, consequências de mutações patogênicas no TRIP4 e ASCC1.
REFERÊNCIAS
1 Davignon, L. et al. The transcription coactivator ASC-1 is a regulator of skeletal myogenesis, and its deficiency causes a novel form of congenital muscle disease, Human Molecular Genetics, 2016; 25(8), 1559-1573.
2 Chaytow H., Huang Y., Gillingwater TH., Faller, KME. The role of survival motor neuron protein (SMN) in protein homeostasis, Cellular and Molecular Life Sciences, 2018; 75(21): 3877-3894.
3 Mendonça, Rodrigo H., et al. "Severe brain involvement in 5q spinal muscular atrophy type 0." Annals of neurology 86.3, 2019: 458-462.
4 Matesanz, Susan E., et al. Clinical course in a patient with spinal muscular atrophy type 0 treated with nusinersen and onasemnogene abeparvovec. Journal of Child Neurology, 2020;35.11: 717-723.
5 Böhm J, et al. Novel ASCC1 mutations causing prenatal-onset muscle weakness with arthrogryposis and congenital bone fractures, Journal of Medical Genetics, 2018; 0:1-5.
6 Oliveira, J., Martins, M., Leite, RP, Sousa, M., Santos, R. The new neuromuscular disease related with defects in the ASC‐1 complex: report of a second case confirms ASCC1 involvement. Clinical Genetics, 2017; 92:434-439.
7 Todd, JE. et al. Next generation sequencing in a large cohort of patients presenting with neuromuscular disease before or at birth. Orphanet Journal of rare diseases, 2015; 10:148.
8 Hunter JM., et al. Review of X-linked syndromes with arthrogryposis or early contractures - aid to diagnosis and pathway identification. American Journal Medical Genetics Part A, 2015; (167A):931-973.
9 Knierim E. et al. Mutations in subunits of the activating signal cointegrator 1 complex are associated with prenatal spinal muscular atrophy and congenital bone fractures. The American Journal of Human Genetics, 2016; (98) 473-489.
10 Torices, S. et al. A truncated variant of ASCC1, a novel inhibitor of NF-KB, is associated with disease severity in patients with rheumatoid arthritis, The Journal of Immunology, 2015; (195): 5415-5420.
11 Chi, B.et al. The neurodegenerative diseases ALS and SMA are linked at the molecular level via the ASC-1 complex, Nucleic Acids Research, 2018; 46 (22) 11939-11951.
12 Giuffrida, MG. et al. A new case of SMABF2 diagnosed in stillbirth expands the prenatal presentation and mutational spectrum of ASCC1, American Journal of Medical Genetics; 2019; 1-5.
13 Lu, W. et al. Novel compound heterozygous pathogenic variants in ASCC1 in a Chinese patient with spinal muscular atrophy with congenital bone fractures 2: Evidence supporting a "Definitive" gene-disease relationship. Molecular Genetics & Genomic Medicine, 2020; 00:e1212.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()