Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(4) - Dezembro 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Apresentação pediátrica da doença de Fabry: sintomas comuns levando ao diagnóstico de uma doença rara
Pediatric presentation of Fabry disease: common symptoms leading to the diagnosis of a rare disease
Graciela S. Rocha1; Mariana D. Ferrari1; Marina V. Balhester1; Osvaldo Theodoro Paz1; Laura Vagnini2; Jacqueline Fonseca3; Zumira Aparecida Carneiro1; Charles Marques Lourenco1; 2; 4
DOI:10.31365/issn.2595-1769.v21i4p211-217
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - ribeirao preto - Please Select - Brasil
2. Centro Paulista de Diagnostico e Pesquisa, Genetica Clínica - RIBEIRAO PRETO - SP - Brasil
3. Laboratório DLE, Genética Bioquímica - ribeirao preto - Please Select - Brasil
4. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
Endereço para correspondência:
Recebido em: 16/11/2020
Aprovado em: 08/05/2021
Instituição: Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - ribeirao preto - Please Select - Brasil
Resumo
A doença de Fabry (DF) é uma doença de origem genética ligada ao cromossomo x. Ocorre devido a um erro inato no metabolismo dos glicoesfigolipídeos (GSL), decorrente da deficiência da enzima alfa-galactosidase A (alfa-Gal), cursando com acúmulo de globotriasilceramida (Gb3). A deficiência dessa enzima ocasiona um acúmulo progressivo de Gb3 dentro do lisossomo, originando complicações em diferentes órgãos e sistemas corporais. Esse armazenamento explica a ocorrência dos primeiros sintomas durante a infância, caracterizados por dor, sensibilidade ao calor e ao frio, doença gastrointestinal, enfatizando a dor abdominal recorrente e a diarreia. Relatamos um caso da DF em uma criança do sexo masculino e ressaltamos a importância dos sinais clínicos e sintomas que conduzem o pediatra ao diagnóstico da doença durante a primeira infância.
Palavras-chave: Doença de Fabry. Erros Inatos do Metabolismo. Cromossomo X. Diagnóstico Precoce
Abstract
Fabry disease (FD) is a disease of genetic origin linked to the x chromosome. It occurs due to an inborn error in the metabolism of glycosphigolipids (GSL), mainly globotriasylceramide (Gb3), secondary to deficiency of the enzyme alpha-galactosidase A (alfa-Gal). The alfa-Gal enzyme deficiency causes progressive accumulation of Gb3 within the lysosome, leading to complications in different organs and body systems. This storage explains the occurrence of the first symptoms during childhood, characterized by pain, sensitivity to heat and cold, gastrointestinal disease, emphasizing recurrent abdominal pain and diarrhea. Here we report a case of FD in a male child and emphasize the importance of the clinical signs and symptoms that the pediatrician gives to the diagnosis of the disease during early childhood.
Keywords: Fabry Disease. Metabolism, Inborn Errors. X Chromosome. Early Diagnosis
INTRODUÇÃO
A doença de Fabry (DF) é uma doença de origem genética ligada ao cromossomo X. Ocorre devido a um erro inato no metabolismo dos glicoesfigolipídeos (GSL), decorrente da deficiência da enzima alfa-galactosidase A (alfa-Gal), cursando com acúmulo de globotriasilceramida (Gb3).1,2 O acúmulo progressivo de Gb3 afeta os sistemas nervoso periférico (acroparestesia) e gastrointestinal (diarreia, dor abdominal após a alimentação, vômitos), olhos (córnea verticillata) e pele (angioqueratoma, hipoidrose ou anidrose).1
Em torno da terceira e quarta década de vida, ocorre um aumento progressivo das lesões cardiovasculares (hipertrofia ventricular esquerda, hipertensão, isquemia miocárdica), renais (insuficiência renal crônica), cerebrais (distúrbios psiquiátricos, acidente vascular cerebral isquêmico e hemorrágico) e o padrão característico da lesão vascular é de pequenos vasos com predileção pelo território vertebro-basilar.1
O processo primário da doença tem início na infância, sobretudo na faixa etária entre 3 e 10 anos em pacientes do sexo masculino e, no sexo feminino, geralmente tem o início mais postergado.3 As mulheres heterozigotas podem ser tão gravemente afetadas como os homens, mas tendem a apresentar a doença de forma mais insidiosa e tardia. Tal fato provavelmente está relacionado com a inativação aleatória do cromossomo X (teoria de Lyon).3 O gene da alfa-Gal, conhecido como GLA, está localizado no cromossomo X (locus Xq22), por isso acomete principalmente homens, embora possa acometer também as mulheres, sendo que a maioria das portadoras heterozigóticas apresenta a doença de forma atenuada.4
O diagnóstico pode ser confirmado através da determinação da atividade da alfa-galactosidase A e do sequenciamento do gene GLA, sendo que a enzima pode ser dosada no plasma, em leucócitos ou pelo método de papel-filtro (do inglês, dried blood spot, DBS).1 A atividade de alfa-Gal, na maioria dos homens afetados, é inferior a 5% do normal,2,3 e em mulheres heterozigóticas podem ocorrer valores de dosagem enzimática dentro do valor de normalidade, exigindo então que o teste genético seja realizado em virtude do mecanismo de inativação randômica do cromossomo X1. Em virtude dessa inativação do cromossomo X (XCI), as mulheres são como um mosaico para a expressão de genes nesse cromossomo, em que há um silenciamento transcricional (aleatório) de um dos cromossomos x em cada célula feminina.5
O tratamento é realizado pela terapia de reposição enzimática, as enzimas recombinantes α-galsidase alfa e α-galsidase beta, ambas são estrutural e funcionalmente semelhantes, sendo administradas via intravenosa a cada 15 dias.1 Existe, recentemente, também a possibilidade do tratamento via oral, através da utilização de chaperona. Tal medicamento atua na ligação da chaperona farmacológica, migalastat, ao sítio ativo da α-galactosidase, gerando a estabilização de certas enzimas mutantes, o que facilita o tráfego adequado para os lisossomos, permitindo que a α-galactosidase catabolize os substratos acumulados.6
O migalastate, administrado via oral, é um tratamento alternativo potencial à terapia de reposição enzimática (TRE) intravenosa, pois o fardo de infusões intravenosas quinzenais por toda a vida pode dificultar a adesão dos pacientes ao tratamento, aumentando assim o risco de lesões de órgãos-alvo.7 Ademais, a TRE pode não atingir os órgãos-alvo (como os rins, coração e cérebro) de maneira tão potente devido à estrutura molecular da enzima recombinante. Já o migalastate, por se tratar de molécula de baixo peso molecular, possui penetração aumentada de órgãos e tecidos. Além disso, teoricamente o acompanhamento de alfa-Gal pelo migalastate aos lisossomos pode mimetizar melhor o tráfego enzimático natural e resultar em atividade de α-Gal mais constante que infusões quinzenais de terapia de reposição enzimática, podendo também ser de administração oral, o que influencia diretamente uma melhor adesão do paciente ao tratamento.7
A DF é uma condição variável com uma progressão insidiosa de sintomas sutis que começam antes do nascimento, visto que estudos histopatológicos neonatais e pré-natais confirmam presença de níveis de Gb3 na placenta e nos tecidos fetais. Esse armazenamento explicaria a ocorrência dos primeiros sintomas durante a infância, principalmente na primeira infância, apresentando dor, sensibilidade ao calor e ao frio, doenças gastrointestinais, especialmente dor abdominal recorrente e diarreia.7
Sendo assim, após a confirmação do diagnóstico da DF, o aconselhamento genético deve ser fornecido ao paciente e seus familiares. É essencial que a família de um paciente portador de DF seja orientada sobre os exames disponíveis e sobre as consequências de um diagnóstico positivo.1
Neste relato, os dados foram obtidos através da revisão do prontuário, após assinatura do termo de consentimento livre e esclarecido. Foram respeitados os princípios éticos de pesquisa com seres humanos, em conformidade com a Resolução n° 466/2012, Resolução n° 510/2016 e que o mesmo obteve aprovação pelo Comitê de Ética em Pesquisa da instituição, sob parecer de número 2.190.929 na data de 28 de julho de 2017 sob o número do CAAE 70623317.7.0000.5581, gerado pela Plataforma Brasil.
RELATO DE CASO
Paciente, sexo masculino, 8 anos de idade, filho de pais jovens e não-consanguíneos, apresentou quadro de dor neuropática sob forma de acroparestesias e intolerância ao calor, sendo encaminhado para avaliação de seus sintomas clínicos.
À história clínica, a mãe do paciente inicia o relato evidenciando os primeiros sintomas observados por volta dos quatro anos de idade do filho, apresentando-se constante ao longo dos anos, sendo eles, dormência, ardência, prurido e edema em região de antebraço e pernas, ocorrências de bolhas e rachaduras em pés, cefaleia em região crânio-frontal e aumento da sensibilidade auditiva. Em suma, no presente momento, o paciente referiu fadiga e dores difusas no corpo, com predomínio em mãos e pés e piora progressiva, impedindo-o de realizar suas tarefas escolares ou atividades físicas. Associada a tais queixas, o mesmo relata episódios esporádicos de intensa cefaleia e de zumbidos, além de dores abdominais e diarreia. Esta última, contudo, referida num período específico da infância com melhora espontânea após hidratação.
Referente aos dados perinatais do paciente, o mesmo nasceu com idade gestacional de 40 semanas e 3 dias, pesando 3,995 quilogramas, 50 cm de comprimento e 35,6 cm de perímetro cefálico. A mãe relatou não ter apresentado nenhuma comorbidade durante o período gestacional. Sobre o desenvolvimento motor, o paciente apresentou sustentação cefálica com seis meses, seguida da sustentação de tronco aos nove meses, sentou sem apoio aos 10 meses, engatinhou aos 11 meses, obteve marcha independente aos 12 meses. Primeiras palavras foram emitidas em torno dos 10 meses e, aos 18 meses, elaborava frases simples.
Não havia queixas, a princípio, quanto às atividades físicas: subia escadas e tolerava bem exercícios; intolerância às atividades físicas e ao calor ficaram mais evidentes a partir dos quatro anos de idade, quando se iniciaram as queixas de acroparestesias. Apesar disso, frequentava escola regular, com aproveitamento, e se encontrava no 3º ano do ciclo básico.
Em sua investigação inicial, realizou exames bioquímicos gerais, inclusive com a dosagem de vitamina B12 sérica, ácido fólico, homocisteína, porém exames revelaram-se dentro da normalidade. Encaminhado para serviço de neuropediatria, fez-se a suspeita de acroparestesia por neuropatia de fibras finas e solicitou-se exame de eletroneuromiografia, que também foi compatível com a normalidade.
Devido à suspeita de síndrome dolorosa da infância sem etiologia definida, foi encaminhado para avaliação em serviço de neurogenética, para investigação de enfermidades que cursassem com dor neuropática (neuropatia de fibras finas) de início precoce.
Ao exame físico, evidenciou-se paciente orientado e lúcido, com fácies atípica com discreta infiltração facial (figura 1), movimentos oculares preservados e simétricos; apresentando força muscular grau 5, reflexos osteotendíneos normoativos; presença de marcha atípica, disestesias em mãos e pés, e eumetria.
Figura 1. Imagem do paciente (uso de imagem autorizada pelos responsáveis com respectiva assinatura de termo de consentimento para utilização de imagens em periódicos).
Fonte: os autores.
Diante dos achados clínicos e laboratoriais, fez-se investigação orientada para a suspeita clínica da DF, a qual foi confirmada a partir de testes enzimáticos e bioquímicos (tabelas 1 e 2).
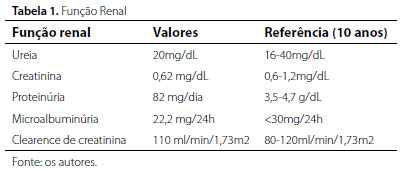
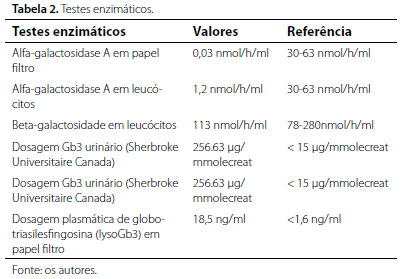
Além dos exames apresentados nas tabelas, foram realizados exames para análise da função cardiológica, como holter de 24h, que evidenciou ritmo sinusal irregular; eletrocardiograma evidenciando sobrecarga de ventrículo esquerdo; ecocardiograma apresentando prolapso e insuficiência de válvula mitral. Também foi realizado o exame oftalmológico do paciente, evidenciando a presença de cornea verticillata. Por fim, verificou-se, por estudo molecular do gene GLA, a presença da mutação c.790C.T, (pAsp264Tyr, pD264Y), confirmando o diagnóstico enzimático da DF.
Em uma breve coleta do histórico familiar, observaram-se outros dados diagnósticos de DF na família materna do paciente, como presença de dor neuropática, insuficiência renal de causa desconhecida, cardiomiopatia hipertrófica, acidente vascular cerebral "criptogênico", sendo possível identificar seu avô e quatro tias, mãe e dois irmãos como também provavelmente afetados por DF (figura 2). O estado de portadoras das mulheres presentes no heredograma (tias do paciente, genitora e irmã) foi confirmado através de análise molecular do gene GLA, e dos homens afetados (avô, primo e irmão), através da análise enzimática da alfa-Gal em leucócitos, com posterior confirmação genética.
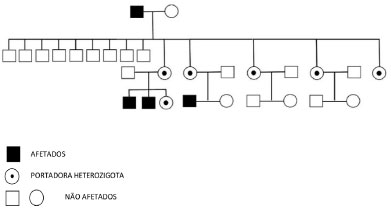
Figura 2. Heredograma da família materna. Fonte: os autores (2021)
Irmã e irmão do paciente apresentavam sintomas semelhantes aos do paciente, exibindo também queixas de acroparestesias e intolerância ao calor, além de diminuição da sudorese. Exames complementares realizados (ecocardiograma, holter de 24h, eletrocardiograma) foram dentro da normalidade, porém ambos os pacientes apresentavam cornea verticillata ao exame oftalmológico com lâmpada de fenda como o caso índex.
Tias do paciente estão em seguimento em setor de clínica médica da cidade de origem. Todas apresentam sintomas e achados clínicos/laboratoriais semelhantes: leve edema em membros inferiores, hipertensão arterial, aumento leve a moderado de proteinúria, hipertrofia de ventrículo esquerdo, alterações de substância branca cerebral (em exame de ressonância magnética cerebral).
DISCUSSÃO
A doença de Fabry (DF) é multifacetada, envolve diversos órgãos e sistemas, o que torna o diagnóstico dessa enfermidade um verdadeiro desafio para o clínico, sendo ainda maior para o médico pediatra. A escassez de dados e de avaliações nos indivíduos diagnosticados no período pré-natal ou durante a infância dificultou a caracterização da história natural da DF na população pediátrica.11 A doença de Fabry pode ser subdiagnosticada devido a sintomas inespecíficos e multiorgânicos.9
Acredita-se que a patogênese da dor na DF está relacionada ao acúmulo lisossomal de Gb3 nos nervos periféricos, nos gânglios da raiz dorsal e na medula espinhal.8 Em particular, a atrofia dos pequenos nervos não mielinizados relacionados à sensação de dor e temperatura estão envolvidos na fisiopatologia da dor neuropática de "fibras finas", acometendo preferencialmente extremidades distais (mãos e pés),12 conforme observado no paciente do relato.
A dor na DF geralmente não responde aos analgésicos convencionais, sendo necessário, na crise álgica, utilizar drogas como meperidina, codeína, morfina ou carbamazepina. Além disso, pacientes com dor crônica podem se beneficiar com fenitoína, amitriptilina ou gabapentina.12,13 Em um estudo com pacientes pediátricos com DF, realizado por Hopkin et al.,13 analisou-se a sintomatologia em 352 doentes com idades variando entre 0-17 anos, com idade média de 12 anos, verificando-se que os sintomas surgiam por volta de 6 anos em meninos e 9 anos em meninas.
No caso relatado, contudo, a idade de aparecimento da dor neuropática foi ainda mais precoce, em torno dos 4 anos de idade, o que também foi observado em estudo de revisão sistemática da DF na infância realizado por Laney et al.,7 em que acroparestesia e dor neuropática surgem, em meninos, entre 2 e 4 anos de idade.
Ademais, o mesmo autor abordou a dor neuropática como sintoma de maior relevância entre os pacientes pediátricos, relatada em 59% dos meninos e 41% das meninas, sendo a idade de início variável de indivíduo para indivíduo, mesmo dentro da mesma família. Usualmente, ela é descrita como ardor e sensação de "alfinetadas e agulhadas", normalmente envolvendo mãos e pés, com irradiação proximal, sendo desencadeada por frio, calor, exercício físico (como o observado no paciente do relato) ou estresse emocional e pode se apresentar como contínua ou episódica.15 Também abordou que tais sintomas não são tipicamente fatais, porém prejudicam claramente a saúde e funcionalidade das crianças afetadas com a doença.13-16
No caso do paciente descrito neste relato, a sintomatologia de dor crônica estava prejudicando seu rendimento escolar, incorrendo em constante absenteísmo. Pacientes pediátricos com DF podem exibir intolerância a extremos de temperaturas. Quando expostos ao calor, podem se queixar de rubor, dor neuropática, dores de cabeça, fadiga ou insolação, fato observado no paciente do relato, inclusive como um fator de exacerbação das crises de dor. Já no frio, pacientes com DF podem apresentar aumento da dor ou dormência nas extremidades.17,18
Sintomas de hipoidrose são marcantes em pacientes com DF, 28,4% de prevalência em pacientes do sexo masculino e 22,2% em pacientes do sexo feminino.14 Embora seja um sintoma aparentemente benigno, a hipoidrose pode levar ao aumento da temperatura corporal em dias mais quentes, o que pode precipitar novas crises de dor, fato observado no paciente do relato com relativa frequência. A dor abdominal é um achado comum na DF, particularmente na faixa etária pediátrica. Geralmente é inespecífica, podendo mimetizar o refluxo gastroesofágico ou uma colite espástica, ocorrendo espontaneamente ou associada a alimentos com alto teor de gordura.19
Diarreia, constipação, náuseas e vômitos também são queixas comuns em pacientes com DF,15 especialmente em crianças menores, embora não tenham sido referidos pela genitora do paciente deste relato como sintomas frequentes. Houve referência, contudo, a esses sintomas durante um período específico da infância do paciente, com melhora espontânea após hidratação, o que pode ou não estar relacionado a sua doença de base.
Já em relação ao sistema oftalmológico, o achado mais frequente da DF é a cornea verticillata, cuja manifestação na infância geralmente não afeta a visão e pode se apresentar previamente a sintomas mais graves. Ademais, Cordeiro et al.1 trazem algumas características típicas ao exame clínico dessa alteração, tais como a dilatação e a tortuosidade vascular, formação de aneurismas em conjuntiva, espirais de córnea com opacidades amareladas irradiando de um ponto próximo ao centro da córnea.
De forma similar, o paciente, apesar de apresentar a típica cornea verticillata, não possuía queixas de baixa acuidade visual.
Um estudo recente com objetivo descritivo epidemiológico sobre a deficiência auditiva e zumbido em crianças com DF concluiu que a perda auditiva e o zumbido são manifestações clínicas bem conhecidas pelos pacientes pediátricos. Todavia, quando realizados os exames de audiometria de alta frequência, uma baixíssima porcentagem dessa população realmente apresentou perda auditiva, afetando 19% dos pacientes.20 Deve-se ressaltar que os indivíduos que apresentavam zumbido possuíam scores gerais de gravidade da doença mais altos. A maioria dos pacientes, em torno da adolescência, relata zumbido, sendo predominantemente meninas. Esses dados sugerem fortemente a importância do acompanhamento audiométrico de todos os indivíduos com DF.20
No caso apresentado, queixas de hiperacusia e zumbido foram relatadas como frequentes, mas a princípio, paciente possuía exame de audiometria dentro da normalidade.
Acerca dos aspectos cardiovasculares da DF, embora poucos pacientes pediátricos apresentem anormalidades cardiovasculares no momento do diagnóstico, o acompanhamento clínico mostra disfunção valvar em 22,6% dos pacientes do sexo masculino. Anormalidades da condução, arritmias e hipertrofia do VE são menos frequentes que as disfunções valvares.14 Um estudo feito por Kampmann et al.17 sobre as manifestações cardíacas em crianças e jovens adolescentes com Fabry relata que, antes da idade adulta, eles podem evoluir com prolapso da válvula mitral e aumento de massa muscular de ventrículo esquerdo.21 Tais mudanças foram observadas independentemente do sexo feminino ou masculino.
De forma similar ao descrito por Kampmann et al.,17 nosso paciente possui eletrocardiograma e ecocardiograma com sobrecarga de ventrículo esquerdo, apresentando, ainda, prolapso e insuficiência de válvula mitral.
Outros achados, como angioqueratomas, são comuns em pacientes com DF, ocorrendo em 14,2% dos pacientes pediátricos, em particular pacientes pré-púberes.14 São pequenas lesões vermelhas não branqueadas, que podem ser encontradas, predominantemente, na virilha, nádegas, abdome e membrana mucosa da boca e lábios.22 Provavelmente devido a sua idade, o paciente do relato ainda não exibe sinais de angioqueratoma.
Entre os achados renais da DF, observa-se que a proteinúria é quase um achado universal em todos os pacientes do sexo masculino e pode começar já aos 10 anos de idade (ou mesmo antes, como foi observado em nosso paciente). Conforme a proteinúria progride, a taxa de filtração glomerular (TFG) diminui progressivamente. A maioria dos pacientes do sexo masculino e 15-20% dos pacientes do sexo feminino desenvolvem doença renal terminal.22 A TRE tem sido defendida em pacientes jovens com DF, especialmente porque seria capaz de deter o dano renal progressivo.10
Ramaswami et al.21 relataram, em estudo observacional de pacientes pediátricos com DF sob terapia de reposição enzimática (TRE), que sintomas gastrointestinais diminuíram em frequência e gravidade em meninos. A dor neuropática, que está significativamente relacionada à qualidade de vida, não apresentou menor prevalência no grupo observado após 1 ou 2 anos de TRE. No entanto, houve redução na prevalência de dor nos pacientes que tiveram crises ou dor crônica no início do estudo. Houve, também, estabilidade do índice de massa ventricular esquerda (MVEi), representando um efeito cardíaco benéfico dessa terapia, uma vez que o aumento da MVEi tem sido demonstrado em crianças sem tratamento com idade semelhante. O paciente certamente se beneficiará da TER, visto que seu diagnóstico foi feito antes que danos irreversíveis a órgãos-alvo como rins e coração fossem estabelecidos.
Em resumo, a DF deve ser considerada em pacientes pediátricos com dor crônica, especialmente em combinação com dor abdominal e outras características (angioqueratomas, hipoidrose e intolerância a atividades físicas). A história familiar de pacientes com doença renal terminal de causa desconhecida ou cardiomiopatia hipertrófica em adultos pode ser uma pista importante para o diagnóstico de DF. O diagnóstico precoce da DF contribui substancialmente não só para o atendimento adequado e acompanhamento dessas crianças, mas também para o aconselhamento genético familiar.
REFERÊNCIAS
1. Cordeiro CA, Oréfice F, Lasmar EP, Santos HH, Valadares ER. Córnea verticilata - marcador clínico da doença de Fabry: relato de caso. Arq Bras Oftalmol. 2007;70(4):701-5. https://doi.org/10.1590/S0004-27492007000400024.
2. Biagini G, Almeida ACSF, Almeida TVR, Silva CAB, Castro BF de, Reche TC, et al. Case report: is low α-Gal enzyme activity suffcient to establish the diagnosis of Fabry disease? J Bras Nefrol. 2017;39(3):333-336. DOI: 10.5935/0101-2800.20170057.
3. Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. DOI: 10.1186/1750-1172-5-30.
4. Blom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JMFG. Recombinant Enzyme Therapy for Fabry Disease: Absence of Editing of Human α-Galactosidase A mRNA. Am J Hum Genet. 2003;72(1):23-31. DOI: 10.1086/345309.
5. Elstein D, Schachamorov E, Beeri R, Altarescu G. X-inactivation in Fabry disease. Gene. 2012;505(2):266-8. DOI: 10.1016/j.gene.2012.06.013. Epub 2012 Jun 16.
6. Germain DP, Hughes DA, Nicholls K, Bichet DG, Giugliani R, Wilcox WR, et al. Treatment of Fabry's Disease with the Pharmacologic Chaperone Migalastat. N Engl J Med. 2016;375(6):545-55. DOI: 10.1056/NEJMoa1510198.
7. Laney DA, Peck DS, Atherton AM, Manwaring LP, Christensen KM, Shankar SP, et al. Fabry disease in infancy and early childhood: a systematic literature review. Genet Med Off J Am Coll Med Genet. 2015;17(5):323-30.
8. Wang RY, Bodamer OA, Watson MS, Wilcox WR; ACMG Work Group on Diagnostic Confirmation of Lysosomal Storage Diseases. Lysosomal storage diseases: diagnostic confirmation and management of presymptomatic individuals. Genet Med. 2011;13(5):457-84. DOI: 10.1097/GIM.0b013e318211a7e1.
9. Rozenfeld PA. Fabry disease: treatment and diagnosis. IUBMB Life. 2009;61(11):1043-1050. DOI:10.1002/iub.257.
10. Prabakaran T, Birn H, Bibby BM, Regeniter A, Sørensen SS, Feldt-Rasmussen U, Nielsen R, Christensen EI. Long-term enzyme replacement therapy is associated with reduced proteinuria and preserved proximal tubular function in women with Fabry disease. Nephrol Dial Transplant. 2014 Mar;29(3):619-25. DOI: 10.1093/ndt/gft452.
11. Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, Lemay R, Tylki-Szymanska A, Wilcox WR. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550-5. DOI: 10.1203/PDR.0b013e318183f132.
12. Ries M, Gupta S, Moore DF, Sachdev V, Quirk JM, Murray GJ, et al. Pediatric Fabry Disease. Pediatrics. 2005;115(3):e344-e355. DOI: https://doi.org/10.1542/peds.2004-1678
13. Ries, M, Ramaswami, U, Parini, R, et al. The early clinical phenotype of Fabry disease: a study on 35 European children and adolescents. Eur J Pediatr. 2003;162:767-772. https://doi.org/10.1007/s00431-003-1299-3
14. Hilz MJ, Stemper B, Kolodny EH. Lower limb cold exposure induces pain and prolonged small fiber dysfunction in Fabry patients. Department of Neurology, New York University, New York, NY, USA. Pain. 2000;84(2-3):361-365 Doi: 10.1016/s0304-3959(99)00236-5
15. Hoffmann B, Reinhardt D, Koletzko B. Effect of enzyme-replacement therapy on gastrointestinal symptoms in Fabry disease. Eur J Gastroenterol Hepatol. 2004;16(10):1067-9. DOI: 10.1097/00042737-200410000-00020
16. Desnick RJ, Brady RO. Fabry disease in childhood. J Pediatr. 2004;144(5 Suppl):S20-6. DOI: 10.1016/j.jpeds.2004.01.051
17. Kampmann C, Wiethoff CM, Whybra C, Baehner FA, Mengel E, Beck M. Cardiac manifestations of Anderson-Fabry disease in children and adolescents. Acta Paediatr. 2008;97:463-469
18. Havranek S, Linhart A, Urbanova Z, Ramaswam U. Early cardiac changes in children with Anderson-Fabry. JIMD Rep. 2013;11:53-64. DOI: 10.1007/8904_2013_222
19. Mehta A, Beck M, Sunder-Plassmann G, Ramaswami U, Parini R, Pintos-Morell G. Natural history and effects of enzyme replacement therapy in children and adolescents with Fabry disease. Oxford PharmaGenesis; 2006. Chapter 31.
20. Najafian B, Svarstad E, Bostad L, Gubler M-C, Tøndel C, Whitley C, et al. Progressive podocyte injury and globotriaosylceramide (GL-3) accumulation in young patients with Fabry disease. Kidney Int. 2011;79(6):663-70. DOI: 10.1038/ki.2010.484.
21. Ramaswami U, Parini R, Pintos-Morell G, Kalkum G, Kampmann C, Beck M, et al. Fabry disease in children and response to enzyme replacement therapy: results from the Fabry Outcome Survey. Clin Genet. 2012;81(5):485-90. DOI: 10.1111/j.1399-0004.2011.01671.x.
22. Morel GP, Beck M. Fabry disease in children and the effects of enzyme replacement. Eur J Pediatr. 2009;168(11):1355-1363. DOI: 10.1007/s00431-009-0937-9
23. Gal A, Hughes DA, Winchester B. Toward a consensus in the laboratory diagnostics of Fabry disease - recommendations of a European expert group. J Inherit Metab Dis. 2011 Apr;34(2):509-14. DOI: 10.1007/s10545-010-9261-9.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()