Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(3) - Dezembro 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Encefalopatia epiléptica por mutação no gene DHX30: Relato do primeiro caso latino-americano
Epileptic encephalopathy by mutation in the DHX30 gene: Report of the first Latin-American patient
Joana Chiara Mecca1; Luciana do Prado Rocha1; Laura Vagnini2; Marcela Almeida1; Patricia Barros Viegas Anno3; Debora de Cassia Tomaz3; Regina Albuquerque3; Jacqueline Fonseca4; Zumira Aparecida Carneiro1; Ana Paula Andrade Hamad5; Fernanda Veiga Gomes6; Charles Marques Lourenco1,2,3
DOI:10.31365/issn.2595-1769.v21i3p164-169
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Centro Paulista de Diagnóstico e Pesquisa, Genética Clínica - Ribeirão Preto - SP - Brasil
3. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
4. Laboratório DLE, Bioquímica Genética - Rio de Janeiro - RJ - Brasil
5. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - Ribeirão Preto - SP - Brasil
6. Instituto Nacional de Saúde da Mulher, Criança e do Adolescente Fernandes Figueira (IFF) - Fiocruz, Neuropediatria - Rio de Janeiro - RJ - Brasil
Endereço para correspondência:
Recebido em: 16/04/2020
Aprovado em: 27/11/2020
Instituição: Faculdade de Medicina - Ribeirão Preto
Resumo
INTRODUÇÃO: Encefalopatia epiléptica por mutação no gene DHX30 (DExH-Box Helicase 30) é uma rara enfermidade genética, caracterizada por déficit intelectual, crises convulsivas e malformações do sistema nervoso central. Pacientes portadores de mutação nesse gene frequentemente apresentam crises convulsivas refratárias a inúmeras terapias medicamentosas, ausência de fala e atraso motor significativo.
OBJETIVO: Relatar primeiro paciente latino-americano afetado por mutações no gene DHX30.
RELATO DE CASO: Paciente do sexo masculino, filho de pais não consanguíneos, encaminhado para investigação de quadro de atraso neuropsicomotor associado a epilepsia de difícil controle. Após extensa investigação e sem obter elucidação diagnóstica, optou-se por realizar sequenciamento completo de exoma, cujo resultado evidenciou mutação de novo no gene DHX30 como causa do quadro clínico do paciente.
DISCUSSÃO: Mutações no gene DHX30 foram recentemente relacionadas ao fenótipo conhecido como neurodevelopmental disorder with severe motor impairment and absent language (NEDMIAL). Trata-se de um espectro clínico que envolve desde sinais de atraso neuropsicomotor com hipotonia a quadros de encefalopatia epiléptica devastadora associados a defeitos de migração neuronal. Através das novas técnicas de sequenciamento de nova geração, como o sequenciamento completo de exoma, novos genes relacionados a distúrbios de neurodesenvolvimento e encefalopatia epiléptica vêm sendo identificados, permitindo a elucidação diagnóstica e aconselhamento genético para famílias afetadas por essas enfermidades.
Palavras-chave: Epilepsia. Deficiência intelectual. Sequenciamento de nucleotídeos em larga escala. Sequenciamento completo do exoma. DHX30.
Abstract
INTRODUCTION: Epileptic encephalopathy due to a mutation in the DHX30 gene (DExH-Box Helicase 30) is a rare genetic disease, characterized by intellectual deficit, seizures and malformations of the central nervous system. Patients with mutations in this gene often have seizures refractory to numerous drug therapies, absence of speech and significant motor delay.
OBJECTIVE: To report the first Latin American patient affected by mutations in the DHX30 gene.
CASE REPORT: Male patient, son of non-consanguineous parents, referred for investigation of neuropsychomotor delay associated with difficult to control epilepsy. After extensive investigation and without obtaining diagnostic clarification, it was decided to perform complete exome sequencing, the result of which revealed a new mutation in the DHX30 gene as the cause of the patient's clinical condition.
DISCUSSION: Mutations in the DHX30 gene have recently been related to the phenotype known as neurodevelopmental disorder with severe motor impairment and absent language (NEDMIAL). It is a clinical spectrum ranging from signs of neuropsychomotor delay with hypotonia to pictures of devastating epileptic encephalopathy associated with neuronal migration defects. Through new generation sequencing techniques, such as complete exome sequencing, new genes related to neurodevelopmental disorders and epileptic encephalopathy have been identified, allowing for diagnostic clarification and genetic counseling for families affected by these diseases.
Keywords: Epilepsy. Intellectual disability. Large-scale nucleotide sequencing. Whole exome sequencing. DHX30.
INTRODUÇÃO
Epilepsia, uma doença comum na infância, apresenta incidência estimada de aproximadamente 70 a cada 100.000 crianças menores de dois anos.1 Diversas etiologias são relacionadas à epilepsia, como infecções, lesões estruturais ou sequela de um acidente vascular cerebral; entretanto, a etiologia genética desponta como causa proeminente de encefalopatia epiléptica de início precoce.2
O surgimento de tecnologias genéticas avançadas de sequenciamento gênico de nova geração, como o sequenciamento do exoma, gerou um significativo aumento na identificação de genes responsáveis por epilepsia na última década. Estima-se que as epilepsias genéticas representam cerca de 30% de todas as síndromes epilépticas.1
As epilepsias genéticas associadas ao atraso de desenvolvimento global e a disfunção cognitiva podem evoluir com um padrão de encefalopatia epiléptica de início precoce, geralmente refratárias ao tratamento com medicações antiepilépticas. Podem ser decorrentes de alterações em genes específicos implicados no processo de transmissão sináptica cerebral, no desenvolvimento e na proliferação neuronal.
O aprofundamento no conhecimento das epilepsias genéticas e seus mecanismos subjacentes permitem desenvolver tratamentos específicos voltados ao genótipo do indivíduo, além do aconselhamento genético da família e o melhor seguimento clínico do paciente.2,4
Recentemente, por meio de técnicas de sequenciamento de nova geração, mutações no gene DHX30 (DExH-Box Helicase 30) foram identificadas em pacientes com encefalopatia epiléptica de início precoce, caracterizando uma nova entidade clínica conhecida como neurodevelopmental disorder with severe motor impairment and absent language (NEDMIAL) (OMIM # 617804).
O gene DHX30 é responsável pela codificação de um conjunto de enzimas, como as helicases, as quais são responsáveis por desenrolar a estrutura do RNA secundário através da hidrólise do ATP,6 expressas nas células neurais na gastrulação.7 Essas células neurais são unidades básicas na construção do sistema nervoso; portanto, caso exista uma mutação nesse gene, haverá repercussão na formação e funcionamento deste sistema.
A origem dessa mutação não é esclarecida, mas se observou que as variantes da proteína DHX30 facilitam a formação de grânulos de stress, os quais funcionam como um gatilho para o início de um processo de inibição da tradução do RNA das células ou até mesmo interferindo na atividade da ATPase. Isso faz com que todos esses processos estejam prejudicados na formação das células neurais e, consequentemente, no desenvolvimento do sistema nervoso.6
O principal objetivo deste estudo é descrever um relato de caso do primeiro paciente latino-americano brasileiro afetado por mutações no gene DHX30 e sua "odisseia diagnóstica". Neste relato, os dados foram obtidos através da revisão do prontuário com a autorização do comitê de ética do hospital e do paciente e por assinatura do termo de consentimento livre e esclarecido.
RELATO DE CASO
Paciente do sexo masculino, quatro anos de idade, filho de casal jovem e não consanguíneo, foi encaminhado para investigação de quadro de atraso neuropsicomotor associado à epilepsia de difícil controle. Nascido de parto cesariana, prematuro, com a idade gestacional de 36 semanas, após gestação sem intercorrências, apresentou peso de nascimento de 2.645g, estatura de 46 cm, perímetro cefálico de 32,5 cm (adequado para idade gestacional) e Apgar 8/10. Evoluiu com icterícia fisiológica, sem necessidade de fototerapia e dificuldade de sucção, necessitando de fórmula infantil.
Em torno do 3º mês de vida, não apresentava sustento de polo cefálico, e ao 6º mês de vida também não possuía controle do tronco. Aos oito meses de vida, apresentava importante atraso neuropsicomotor, e nessa ocasião foi realizada a primeira ressonância nuclear magnética (RNM) de encéfalo, dentro dos padrões da normalidade. Foi indicado acompanhamento multidisciplinar com terapia ocupacional e fisioterapia. Durante as sessões, notaram-se crises disperceptivas de parada comportamental em uma média de oito vezes ao dia. Aos 12 meses, realizou eletroencefalograma em sono e vigília, evidenciando lentificação moderada da atividade de base e ondas aguda na região frontocentral à direita, confirmando o diagnóstico de epilepsia focal.
Mesmo após a introdução do divalproato de sódio, o paciente evoluiu sem controle das crises epilépticas e apresentou efeito colateral de alteração de humor e do comportamento. O padrão de crises referido era de raros episódios de crises motoras tônicas e gelásticas, associados a crises disperceptivas com frequência média de 40 ao dia. Um novo eletroencefalograma aos 22 meses mantinha a lentificação da atividade de base e ondas lentas na região frontal esquerda. Utilizou inúmeras medicações antiepilépticas como Fenobarbital, Clonazepam, Lamotrigina, Carbamazepina, Topiramato e Levetiracetam, sem melhora efetiva das crises e com efeitos colaterais associados. Aos dois anos, nova RNM de encéfalo evidenciou sinais de atrofia cerebral difusa, mais evidente nos lobos temporais e hipocampo (figura 1).
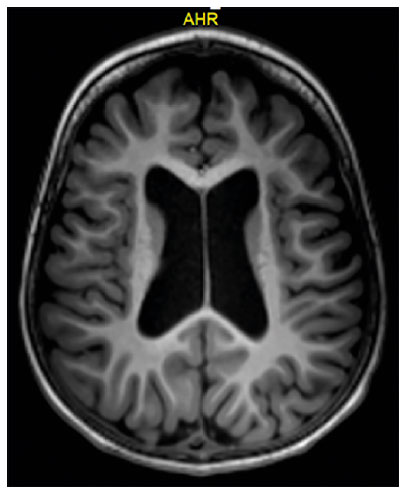
Figura 1. Atrofia cerebral cortical de predomínio frontal, em RNM de encéfalo aos 25 meses de vida.
Em decorrência do quadro de refratariedade de crises epilépticas, o paciente foi encaminhado a serviço terciário especializado no tratamento de epilepsia; não houve, entretanto, indicação cirúrgica, sendo preconizada apenas modificação do tratamento medicamentoso.
Encaminhado ao serviço de neurogenética, realizou investigação com o exame de comparative genomic hybridization (CGH)-Array10 cujo resultado foi normal, excluindo microdeleções e microduplicações cromossômicas. Realizado painel genético para investigação de epilepsia de início precoce, abrangendo cerca de 50 genes utilizando a tecnologia de sequenciamento de nova geração sem anormalidades (Anexo I). Por fim, aos três anos e dois meses de idade, realizou o sequenciamento completo do exoma com identificação da variante patogênica Chr3:47.887.953 A>G (p.Thr425Ala) no gene DHX30, não evidente nos seus genitores, sendo, portanto, uma variante de novo.
Atualmente, o probando apresenta atraso no desenvolvimento motor, hipotonia axial e apendicular, ausência de marcha, afasia, distúrbio do sono, hipermobilidade articular com frouxidão ligamentar, estrabismo, crises epilépticas e dificuldade de alimentação, sendo um quadro compatível com os demais casos da literatura. A família optou por suspender todos os medicamentos antiepilépticos com a justificativa de ausência de resposta clínica.
DISCUSSÃO
O conhecimento sobre a fisiopatogênese das encefalopatias epilépticas tem enriquecido com os avanços após a introdução das tecnologias de "sequenciamento de nova geração" (NGS, do inglês next generation sequencing), particularmente, do sequenciamento completo de exoma. Este método se tornou uma ferramenta de investigação para casos de pacientes que possuem critérios clínicos de encefalopatia epiléptica precoce, que geralmente é refratária ao tratamento convencional e inicia-se nos primeiros anos de vida.2
Temos avançado na identificação de genes de encefalopatias epilépticas, porém o acesso a essa investigação ainda é difícil, retardando ou até impossibilitando o diagnóstico genético. Com a introdução das técnicas de sequenciamento de nova geração, tornou-se possível sequenciar vários genes simultaneamente. Entre um dos genes descobertos pelo avanço das técnicas moleculares, podemos destacar o Deah-Box polypeptide 30 (DExH-Box Helicase 30) (gene DHX30) (OMIM 616423), cuja identificação só foi possível a partir de estudos de sequenciamento dos exons utilizando as técnicas de sequenciamento de nova geração.2
Recentemente, as mutações no gene DHX30 foram descritas como uma rara causa de encefalopatia epilética e estão associadas a alterações cognitivas e comportamentais. Esse gene pertence a uma família de enzimas conhecidas como helicases, que usam a hidrólise do ATP para desenrolar a estrutura do RNA secundário,6 expressa nas células neurais durante a gastrulação.7 Apesar de ser uma mutação em células neurais em uma fase precoce da embriogênese, nenhum dos pacientes descritos até o momento, com exceção de um caso, apresentou malformações do sistema nervoso central ou defeitos de migração neuronal.
Os pacientes portadores de variantes patogênicas nesse gene apresentam uma clínica compatível com encefalopatia epiléptica, caracterizada por deficiência intelectual, afasia, atraso motor significativo, crises epilépticas e alterações na neuroimagem com atrofia cerebral e atraso de mielinização.6
É desconhecido como alterações nesse gene podem levar ao fenótipo de encefalopatia epiléptica. Em estudos experimentais realizados em camundongos, mutações em homozigose no gene DHX30 levam à letalidade, enquanto os camundongos heterozigotos são aparentemente normais e férteis em um primeiro momento. Alguns estudos ainda sugerem que a superexpressão do gene DHX30 pode inibir o RNA do vírus e, assim, reduzir a capacidade de infecção do HIV-1,we identified a novel gene called helicase family gene related to gastrulation (helG7,8 demostrando o quanto ainda se desconhece sobre as funções desse gene.
Estudo recente descreveu 12 casos de encefalopatia epiléptica, os quais se assemelharam ao paciente deste relato.6 O probando reproduz o fenótipo descrito nos pacientes do estudo, apresentando deficiência intelectual, afasia, dificuldade de alimentação, características autistas, distúrbios do sono, atraso do desenvolvimento motor, hipotonia muscular e presença da mutação no gene DHX30 (tabela 1).

Anormalidades na RNM, como redução volumétrica cerebral difusa e atraso de mielinização, estão presentes na maior parte dos casos estudados como consequência da encefalopatia epiléptica, assim como o estrabismo e a hipermobilidade articular, presentes também no paciente relatado.6
A capacidade de deambulação está presente em aproximadamente metade dos pacientes estudados, mas estava ausente no probando. Crises epilépticas estão presentes na minoria dos casos descritos na literatura, porém, merece destaque no paciente deste relato, visto que apresenta elevado número de episódios diários e refratariedade medicamentosa.
Apesar de se tratar de quadros fenotipicamente semelhantes, aparentemente não há uma mutação comum a todos os pacientes. O probando apresenta uma variante p.Thr425Ala que não está presente em nenhum dos 12 pacientes identificados até o momento. Todos os pacientes relatados até hoje eram casos de mutações de novo, ou seja, não herdada de seus genitores.6 Isso tem implicações no aconselhamento genético, visto que a recorrência de mutação desse gene em outra gestação seria extremamente baixa, menos de 1%.3
CONSIDERAÇÕES FINAIS
Relatamos o primeiro caso de um paciente latino-americano com mutações no gene DHX30, corroborando o que a literatura previamente apresentou e definindo a síndrome da mutação DHX30 - síndrome NEDMIAL - como uma doença caracterizada por encefalopatia epiléptica, atraso global de desenvolvimento, atraso da fala e deficiência intelectual. Além disso, ressaltamos que a mutação apresentada pelo probando, p.Thr425Ala, nunca foi relatada previamente.
REFERÊNCIAS
1. Orsini A, Zara F, Striano P. Recent advances in epilepsy genetics. Neurosc Lett. 2018; 667: 4-9.
2. Myers KA, Johnstone DL, Dyment DA. Epilepsy genetics: Current knowledge, applications, and future directions. Clin Genet. 2019; 95:95-111.
3. Møller RS, Dahl HA, Helbig I. The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn. 2015;15(12):1531-1538.
4. Patel J, Mercimek-Mahmutoglu S. Epileptic Encephalopathy in Childhood: A Stepwise Approach for Identification of Underlying Genetic Causes. Indian J Pediatr. 2016; 83(10): 1164-1174.
5. Eldomery MK, Coban-AkdemirZ, Harel T, et al. Lessons learned from additional research analyses of unsolved clinical exome cases. Genome Med. 2017;9:1-15.
6. Lessel D, Schob C, Küry S, et al. De Novo Missense Mutations in DHX30 Impair Global Translation and Cause a Neurodevelopmental Disorder. Am J Hum Genet. 2017;101:716-724.
7. Zheng HJ, Tsukahara M, Liu E, et al. The novel helicase helG (DHX30) is expressed during gastrulation in mice and has a structure similar to a human DExH box helicase. Stem Cells Dev. 2015; 24(3):372-383.
8. Ye P, Liu S, Zhu Y, Chen G, Gao G. DEXH-Box protein DHX30 is required for optimal function of the zinc-finger antiviral protein. Protein Cell. 2010;1:956-964.
9. Mercimek-Mahmutoglu S, Patel J, Cordeiro D, et al. Diagnostic yield of genetic testing in epileptic encephalopathy in childhood. Epilepsia. 2015;56(5): 707-716.
10. Albertson DG, Pinkel D. Genomic microarrays in human genetic disease and cancer. Hum Mol Genet. 2003;12 Spec No 2:R145-52.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()