Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(3) - Dezembro 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Otavio Miranda
- Maria Felicia Avelino
- Larissa Bissoli
- Maria Carolina Antunes Oliveira
- Laura Vagnini
- Jacqueline Fonseca
- Zumira Aparecida Carneiro
- Regina Albuquerque
- Patricia Barros Viegas Anno
- Ana Paula Andrade Hamad
- Iabela Guimarães
- Juliana Maria Faccioli Sicchieri
- Marcela Almeida
- Charles Marques Lourenco
Relato de Caso
Avaliação neuropsicológica em paciente adolescente com fenilcetonúria e diabetes mellitus tipo 1: Relato de caso
Neuropsychological evaluation in a teenager patient with phenylketonuria and type 1 diabetes mellitus: a case report
Otavio Miranda1; Maria Felicia Avelino1; Larissa Bissoli1; Maria Carolina Antunes Oliveira1; Laura Vagnini2; Jacqueline Fonseca3; Zumira Aparecida Carneiro1; Regina Albuquerque4; Patricia Barros Viegas Anno4; Ana Paula Andrade Hamad5; Iabela Guimarães4; Juliana Maria Faccioli Sicchieri6; Marcela Almeida1; Charles Marques Lourenco1,2
DOI:10.31365/issn.2595-1769.v21i3p170-174
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Centro Paulista de Diagnóstico e Pesquisa, Geneéica Clínica - Ribeirão Preto - SP - Brasil
3. Laboratório DLE, Bioquímica Genética - Rio de Janeiro - RJ - Brasil
4. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
5. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - RIBEIRAO PRETO - SP - Brasil
6. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Medicina Interna - RIBEIRAO PRETO - SP - Brasil
Endereço para correspondência:
Recebido em: 05/03/2020
Aprovado em: 14/01/2021
Instituição: Faculdade de Medicina - Ribeirão Preto
Resumo
A fenilcetonúria (PKU) é uma aminoacidopatia que cursa com hiperfenilalaninemia decorrente de disfunção ou ausência da enzima fenilalanina hidroxilase (PAH), levando ao acúmulo de fenilalanina plasmática, acarretando comprometimento do desenvolvimento do sistema nervoso central sob a forma de deficiência intelectual, microcefalia, transtornos de humor e de comportamento. O tratamento deve ser iniciado o mais precocemente possível visando à neuroproteção e consiste, basicamente, em dietoterapia com rigorosa restrição da ingestão de proteínas e uso de uma fórmula de aminoácidos especial isenta ou com baixos teores de fenilalanina. Estudos recentes mostram a importância do tratamento dietético por toda a vida do paciente fenilcetonúrico, pois mesmo após o desenvolvimento neural, altas concentrações de fenilalanina podem promover alterações das funções cognitivas e desmielinização cerebral. Este artigo apresenta caso clínico de paciente adolescente com PKU, porém em uso irregular da fórmula, apresentando sintomas de déficit cognitivos comprovados pelo exame neuropsicológico (Escala Wechsler Abreviada de Inteligência - WASI) associados à coexistência de diabetes mellitus do tipo I como comorbidade.
Palavras-chave: Fenilcetonúrias. Fenilalanina hidroxilase. Fenilalanina. Dietoterapia. Testes neuropsicológicos.
Abstract
Phenylketonuria (PKU) is an aminoacidopathy which occurs with hyperphenylalaninemia resulting from dysfunction or absence of phenylalanine hydroxylase (PAH). Accumulation of phenylalanine in plasma compromises the development of the central nervous system. Consequently, the clinical condition of patients encompasses intellectual disability, microcephaly, and mood and behavior disorders. Treatment must be initiated immediately for neuroprotection and consists in dietotherapty with strict restriction of ingestion of FAL and the use of an amino acid special formula with lower levels or absence of phenylalanine. Recent studies attest the importance of dietetic treatment throughout the life of patients with PKU, as concentrations of phenylalanine may promote alterations in cognitive functions and brain demyeliation. This article presents a female teenager patient with PKU who irregularly takes amino acids formula showing symptoms of cognitive deficit confirmed by neuropsychological test (Wechsler Abbreviated Scale of Intelligence - WASI) and the coexistence of type 1 diabetes mellitus, making her dietotherapy even more complex.
Keywords: Phenylketonurias. Phenylalanine hydroxylase. Phenylalanine. Diet therapy. Neuropsychological tests.
INTRODUÇÃO
A fenilcetonúria (PKU; OMIM 261600) é uma aminoacidopatia, ou seja, é um erro inato do metabolismo (o mais frequente deles), mais especificamente de aminoácidos, e cursa com hiperfenilalaninemia. Esta entidade clínica tem uma prevalência global de 1:10.000 recém-nascidos, sendo que sua incidência varia étnica e geograficamente. As maiores incidências são encontradas na Turquia (1:2.600) e na Irlanda (1:4.500). No Brasil, a prevalência, em 2003, era de aproximadamente 1:24.000.1,2,3
A PKU é caracterizada por um defeito na produção ou na função da enzima Fenilalanina hidroxilase (PAH). O gene da PAH (responsável por 98% das causas de hiperfenilalaninemia) encontra-se no locus 12q22-24.1, estende-se por 90 Kb e possui mais de 500 mutações identificadas, gerando um amplo espectro de fenótipos4.
A PAH é uma enzima hepática dependente de ferro (não-heme), de tetraidrobiopterina (BH4) e oxigênio molecular, sendo responsável pela hidroxilação da L-fenilalanina convertendo-a em L-tirosina. Sua deficiência cursa com um acúmulo de fenilalanina (FAL) e consequente ativação de uma via alternativa de metabolização da mesma, fazendo com que seus metabólitos gerados (especialmente o ácido fenilpirúvico e a fenilacetilglutamina) se acumulem nos tecidos e sejam eliminados na urina, deixando-a com um odor característico - fato este que origina o nome da doença2,4.
Além disso, os níveis elevados de fenilalanina e seus derivados dificulta a mielinização do SNC, alteram a síntese de neurotransmissores e comprometem o desenvolvimento sináptico e dendrítico5. Em decorrência, o quadro clínico desses pacientes envolve deficiência intelectual, microcefalia, transtornos de humor e de comportamento6.
O BH4 é um cofator enzimático necessário para a atividade da PAH e sua deficiência é a causa de 2% dos casos de hiperfenilalaninemia7. O diagnóstico da PKU, normalmente, é feito com a dosagem de fenilalanina no teste de sangue do calcanhar de recém-nascidos. No Brasil, esse exame está incluso no "teste do pezinho" e é um direito garantido a todo recém-nascido pela Portaria no 822/GM, de 06 de junho de 20018. Os valores de fenilalanina considerados normais devem estar abaixo de 4 mg/dL. Quando superiores, mesmo com as divergências entre as classificações de hiperfenilalaninemia, o tratamento dietético já está indicado.5,6
O tratamento visa à neuroproteção e deve ser iniciado, preferencialmente, antes das primeiras duas semanas de vida e continuar durante toda a vida do paciente.9 Classicamente, o tratamento consiste em dietoterapia com rigorosa restrição da ingestão de FAL. Alimentos como carne, leite e seus derivados, ovos, leguminosas, e alimentos que contêm aspartame são contraindicados nesta dieta. O consumo livre de alimentos fica restrito aos açúcares, óleos vegetais e algumas frutas cítricas.5,6,8,9
Além da dieta, é necessário o uso de uma fórmula de aminoácidos especial isenta ou com baixos teores de fenilalanina, suplementada com tirosina, selênio, vitaminas e minerais. Essa fórmula fornece cerca de 80% da proteína dietética diária para o adequado desenvolvimento e crescimento dos fenilcetonúricos, e deve ser manejada individualmente de acordo com a idade, atividade enzimática residual, estado de saúde e velocidade de crescimento desses pacientes.7,10
Outrora, pensava-se que a dietoterapia fosse necessária somente no período da infância, mas estudos recentes mostram a importância do tratamento dietético por toda a vida dos pacientes com diagnóstico de fenilcetonúria, pois mesmo após o completo desenvolvimento neural, altas concentrações de fenilalanina podem promover alterações das funções cognitivas, sujeitando o doente às consequências da hiperfenilalaninemia.9,10
A busca por novas alternativas e terapias adjuvantes tornou-se necessária, visto que pacientes adultos diagnosticados com fenilcetonúria têm maior dificuldade em manter a dieta restritiva. Consequentemente, há um aumento da fenilalanina que, como já citado, apesar de não causar deficiência intelectual, cursa com o comprometimento de funções executivas e desmielinização cerebral.6,8
No caso da PKU, cerca de 75% dos pacientes portadores da doença e não tratados apresentam algum tipo de alteração neurológica.21 Tais alterações decorrem principalmente porque o excesso de fenilalanina compromete a mielinização, assim como a síntese de neurotransmissores dependentes da dopamina.22 Os neurônios dopaminérgicos do córtex pré-frontal são particularmente sensíveis à diminuição de tirosina circulante.22 A maioria dos estudos que avaliou o perfil cognitivo de pacientes com PKU encontrou poucas diferenças entre medidas gerais de QI destes pacientes com seus pares saudáveis. As principais alterações cognitivas encontradas foram em medidas de funções executivas.21,22,23,,24 Tais funções são sabidamente associadas ao córtex pré-frontal.
Neste estudo será relatado o caso de uma paciente com PKU que também foi diagnosticada com diabetes mellitus tipo I (DM I). O DM também é uma condição metabólica que pode cursar com alterações cognitivas, principalmente em atenção, memória e funções executivas.25,26,27
RELATO DE CASO
Paciente do sexo feminino, 18 anos, filha de casal jovem e não-consanguíneo, foi encaminhada para avaliação de controle de níveis de fenilalanina. Nascida de parto cesáreo, a termo, sem intercorrências gestacionais, pesando 3.600g, comprimento de 45cm, apresentou icterícia no terceiro dia de vida, recebendo alta com quatro dias de vida. Após realização de teste de triagem neonatal, paciente foi diagnosticada com forma clássica de fenilcetonúria ("PKU"), iniciando terapia com restrição de fenilalanina e uso de fórmula designada para pacientes com PKU.
Marcos de neurodesenvolvimento foram considerados dentro da normalidade (ficou em pé sem apoio aos 8 meses, andou com 12 meses, montava frases com 1ano 6 meses). Cursa, atualmente o ensino médio, apresentando dificuldades de concentração e acompanhamento das aulas, tendo sido reprovada no 3º ano do ensino fundamental e no 1º ano do ensino médio.
Paciente, desde o primeiro ano de vida, faz dieta hipoproteica suplementada com fórmula de aminoácidos isenta de fenilalanina (Control PKU3 Plus®); vem, porém, há alguns anos em uso irregular da fórmula. Sintomas de déficit de atenção e de alteração de função executiva estavam presentes na paciente e ficaram acentuadas ao longo dos anos.
Aos 18 anos, realizou os primeiros exames neuropsicológicos: Escala Wechsler Abreviada de Inteligência (WASI), evidenciou QI de 83 (dentro da normalidade, na faixa média inferior); avaliação de função executiva, que foi compatível com prejuízos na capacidade de formação de estratégia e na flexibilidade cognitiva, além de desempenho comprometido tanto em tarefa de fluência verbal fonológica quanto em tarefa de fluência verbal semântica. Portanto, seu perfil neuropsicológico após testes iniciais, em suma, indicou comprometimentos múltiplos, abrangendo diversas funções cognitivas.
De antecedentes mórbidos, paciente apresentou dois episódios de convulsões (aos três anos e meio e o aos cinco anos de idade). Iniciou-se terapia com fenobarbital que, posteriormente, foi suspensa e a paciente não apresentou novas crises.
Aos 13 anos, abriu quadro de diabetes mellitus tipo 1 (DM1) e iniciou terapia insulínica associada à dieta hipocalórica e restrita em carboidratos. No curso desse tratamento, paciente refere duas internações decorrentes de cetoacidose: a primeira com 15 anos de idade e a segunda com 18 anos. Duas semanas após a última internação, aos 17 anos, foi diagnosticada com neuropatia diabética em membros inferiores e superiores.
Atualmente, segue com uso de insulina (80UI/dia), pregabalina (150 mg/dia), levonogestrel (0,15mg/dia), etinilestradiol (0,03mg/dia) e, esporadicamente, amitriptilina, conforme prescrição médica.
Paciente compareceu em consulta em bom estado geral, colaborativa, mas em estado nutricional deficitário apresentando índice de massa corporal (IMC) de resultado 17,8 (baixo-peso) e com queixas de dores em membros. Ao exame físico: ausência de dismorfismos faciais; face simétrica com mímica preservada; movimentos oculares sem anormalidade; fígado e baço impalpáveis e ausência de dor e massa em palpações superficial e profunda; presença de disestesias dolorosas (em "queimação") em membros inferiores e superiores; reflexos e sensibilidade preservados; ausência de sinais de irritação meníngea; marcha atípica.
DISCUSSÃO
A fenilcetonúria (PKU) tornou-se um paradigma de doença metabólica hereditária passível de intervenção terapêutica no campo da genética clínica, justificando, inclusive, ter sido a doença que impulsionou o surgimento da triagem neonatal. Na PKU, devido à ineficiente atividade da enzima FAL, há um bloqueio na conversão da fenilalanina em tirosina. Sem o tratamento adequado, os níveis plasmáticos de fenilalanina se elevam e, consequentemente, geram efeitos neurotóxicos que resultam em anormalidades estruturais e grave deficiência intelectual.12
Existem duas hipóteses em relação aos danos neurológicos gerados pela PKU não tratada: enquanto a primeira sugere uma relação entre a baixa concentração plasmática de tirosina e o dano cerebral, a segunda sugere que o fenótipo observado nesses pacientes seja decorrente de hipomielinização de áreas específicas do cortex cerebral.12
No Brasil, o teste do pezinho é realizado desde a década de 70, mas o Programa Nacional de Triagem Neonatal (PNTN), que tornou o teste obrigatório, foi instituído através da Portaria GM/MS no 822, em 6 de junho de 2001.5 O objetivo do programa é ampliar a cobertura de realização do teste e, com isso, tornar mais rápido e preciso o diagnóstico de doenças metabólicas inatas, entre elas, a fenilcetonúria.13 Com o diagnóstico e o tratamento precoces, o prognóstico do paciente é melhor e o risco de danos neurológicos é reduzido.14 No entanto, o portador de fenilcetonúria ainda tem sua pontuação de QI abaixo da média dos indivíduos saudáveis.15
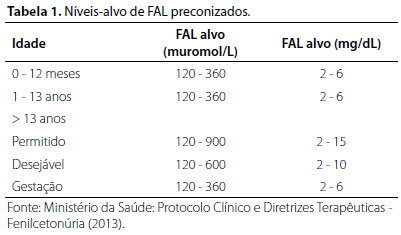
Atualmente, sabe-se que o tratamento deve ser iniciado, idealmente, até o décimo dia de vida e mantido ao longo de toda a vida do paciente, sendo que este consiste em dietoterapia com baixa ingestão de proteínas com a finalidade de evitar o aumento da concentração plasmática de fenilalanina.15,16 O alvo terapêutico da concentração plasmática de fenilalanina, de acordo com o Protocolo Clínico e Diretrizes Terapêuticas para tratamento da Fenilcetonúria do Ministério da Saúde encontra-se na tabela 1.16
Ponto importante a ser considerado nesses pacientes é o impacto psicossocial que a doença gera tanto no indivíduo quanto em sua família.11,12 Como já citado, pacientes adultos têm maior dificuldade em manter a dieta restritiva devido ao planejamento prévio exigido, odor e sabor desagradáveis da fórmula e consequente perda de qualidade de vida.11 Como consequência, observa-se a associação entre PKU e transtornos internalizantes, depressão e ansiedade.11,12,17
Em relação à população normal, a incidência de transtornos gerais de humor nos pacientes fenilcetonúricos não é significantemente maior; observa-se, porém, um padrão nos transtornos mais prevalentes. Em geral, esses pacientes apresentam transtornos internalizantes, que são caracterizados por tristeza, retraimento, queixas somáticas e medo.12,17 No entanto, mesmo nos pacientes que iniciarem precocemente a dietoterapia, alguns achados clínicos poderão estar presentes, por exemplo: redução de QI, déficit de atenção, baixa autoestima, fobias e transtorno de ansiedade e hiperatividade.19
O DM1 é consequência de um processo autoimune mediado pelos linfócitos T, que atuam nas células-beta pancreáticas, levando à diminuição da produção de insulina.
Um paciente com DM1 necessita de insulina, monitorização glicêmica e equilíbrio entre a ingestão de carboidratos e atividade física para ter melhor qualidade de vida e evitar complicações que poderão evoluir para óbito. Ainda assim, algumas vezes, esse conjunto de medidas terapêuticas pode não ser suficiente e poderão ser propostos os transplantes de pâncreas ou de ilhotas, cada um com seus critérios de inclusão.18 Atualmente, outras opções tratamentos estão sendo desenvolvidas, com o objetivo de modular a resposta autoimune18.
Dentre as possíveis complicações do DM, observam-se: vasculopatia, retinopatia, neuropatia periférica, eventos hipoglicêmicos (devido ao uso inadequado de insulina exógena) e alterações cognitivas, como sugerem alguns estudos.18
Os principais fatores de risco para alteração cognitiva relacionada ao DM1 são: idade precoce ao diagnóstico e "cronicidade" da doença (tempo sob o quadro de DM)21.
Na paciente do relato, observam-se complicações associadas ao quadro de DM1, como a neuropatia periférica e episódios de internação por piora de cetoacidose. O mau controle do quadro de diabetes também pode ser correlacionado ao fato de, por conta das restrições dietéticas associadas ao PKU, a paciente ter marcante dificuldade de estabelecer uma rotina alimentar que possa ser compatível com ambas as doenças. Ademais, em virtude do seu quadro neurocognitivo, a paciente apresenta dificuldades de função executiva que podem se correlacionar com maior dificuldade de adesão à dietoterapia, contribuindo ainda mais para o descontrole de níveis glicêmicos e de fenilalanina.11
Esse caso é notável porque a paciente apresenta ambas as doenças que, mesmo não tendo baixas prevalências na população mundial, raramente coexistem no mesmo indivíduo. A associação dessas patologias exerce grande impacto na qualidade de vida do paciente, pois, como já citado, a fenilcetonúria contribui negativamente para o quadro neuropsicológico do paciente e o DM1 restringe ainda mais a dieta do indivíduo.11,17,18
O caso em questão evidencia também a importância da avaliação neuropsicológica em pacientes adultos com PKU não só para diagnóstico de quadros neuropsiquiátricos associados à doença de base, mas também no seguimento de pacientes que apresentam dificuldade de adesão à dieta em PKU ou passem a utilizar terapia medicamentosa específica (como a sapropterina), com o objetivo de obter liberalização da dieta hipoproteica.20
REFERÊNCIAS
1. Omin® and Online Mendelian Inheritance in Man [online]. OMIM Gene Map - Cromossoma: 12. [acesso em 22 set. 2018]. Disponível em https://www.omim.org/geneMap/12?start=1&limit=10&highlight=6677.
2. Dos Santos MP, Haack A. Fenilcetonúria: diagnóstico e tratamento. Revista Brasileira de Ciência e Saúde [online] 2012 novembro. [acesso em 10 mar. 2019]; 23 (263-270). Disponível em http://bvsms.saude.gov.br/bvs/artigos/fenilcetonuria_diagnostico_tratamento.pdf
3. Monteiro LTB, Cândido LMB. Fenilcetonúria no Brasil: evolução e casos. Revista de nutrição [online] 2006 maio-junho. [acesso em 22 set. 2018]; 19 (381-387). Disponível em http://www.academia.edu/5171554/Fenilceton%C3%BAria_no_Brasil_evolu%C3%A7%C3%A3o_e_casos
4. Ceolato JC. Análise molecular do gene pah em pacientes com fenilcetonúria e uma abordagem estrutural da enzima fenilalanina hidroxilase. Porto Alegre: Centro de Biotecnologia da UFRGS, 2011 Dissertação de Mestrado de Pós-graduação em Biologia Celular e Molecular da UFRGS.
5. Kyriakie S, Hoffmann GF, Karl RS. Phenylketonuria. In Pediatric Endocrinology and Inborn Errors of Metabolism. 2 ed. [eBook]. New York, USA: McGraw-Hill Education; Maio de 2017. [acesso em 18 out. 2018]. Disponível em https://accesspediatrics.mhmedical.com/content.aspx?bookid=2042§ionid=154110854
6. Scriver CR, Beaudet AL, Valle D, Sly W S, Childs B, Kinzler KW, Vogelstein B. The metabolic and molecular bases of inherited disease. 8 ed. [eBook]. New York: McGraw-Hill Education; 2001. [acesso em 18 out. 2018]. Disponível em https://ommbid.mhmedical.com/content.aspx?bookid=971§ionid=62673211
7. Bracher A, Eisenreich W, Schramek N, Ritz H, Götze E, Herrmann A, Gütlich M. Biosynthesis of pteridines: NMR studies on the reaction mechanisms of GTP cyclohydrolase I, pyruvoyltetrahydropterin synthase, and sepiapterin reductase. The Journal of Biological Chemistry [online] 1998 outubro. [acesso em 29 set. 2018]; 273 (28132-28141). Disponível em https://www.ncbi.nlm.nih.gov/pubmed/9774432
8. Brasil. Portaria SAS/ MS n° 712, de 17 de dezembro de 2010. Protocolo Clínico e Diretrizes terapêuticas - Fenilcetonúria. Diário Oficial da União 22 dez. 2010. Edição 244: seção 1; p. 107.
9. Jurecki ER, Cederbaum S, Kopesky J, Perry K, Rohr F, Sanchez-Valle A, Viau KS, Sheinin MEU, Cohen-Pfeffer JL. Adherence to clinic recommendations among patients with phenylketonuria in the United States. Mol. Genet Metab. 3, [online] 2017 jan. [acesso em 19 out. 2018]; 120: 190-197. Disponível em https://www.sciencedirect.com/science/article/pii/S1096719217300057?via%3Dihub
10. National Institutes of Health Consensus Development Panel. Phenylketonuria: screening and management. Pediatrics, Elk Grove Village [online] 2001 out. [acesso em 13 nov. 2018] 108 (4): 972-982. Disponível em https://www.ncbi.nlm.nih.gov/pubmed/11581453
11. Levy HL, Milanowski A, Chakrapani A, Cleary M, Lee P, Trefz FK, Whitley CB, Feillet F, Feigenbaum AS, JD Bebchuk, Christ-Schmidt H, Dorenbaum A. Efficacy of sapropterin dihydrochloride (tetrahydrobiopterin, 6R-BH4) for reduction of phenylalanine concentration in patients with phenylketonuria: a phase III randomised placebo-controlled study. The Lancet [revista em internet] 2011 ago. [acesso em 13 nov. 2018];370(9586):504-510. Disponível em https://www.ncbi.nlm.nih.gov/pubmed/17693179
12. Gentile JK, Ten Hoedt AE, Bosch AM. Psychosocial aspects of PKU: Hidden disabilities - A review. Mol Genet Metab. [online] 2009 out. [acesso em 19 mar. 2019];99:S64-7. Disponível em https://www.ncbi.nlm.nih.gov/pubmed/20123473
13. Moreira M. "Teste do Pezinho" completa 10 anos no Brasil. Ciênc saúde coletiva. [online] 2011. [acesso em 19 mar. 2019]; 16:26-7. Disponível em http://www.scielo.br/scielo.php?script=sci_arttext_pr&pid=S1413-81232011010200001
14. Anastasoaie V, Kurzius L, Forbes P, Waisbren S. Stability of blood phenylalanine levels and IQ in children with phenylketonuria. Mol Genet Metab. [online] 2008 setembro-outubro; [acesso em 19 mar. 2019]; 95(1-2):17-20. Disponível em https://www.ncbi.nlm.nih.gov/pubmed/18703366
15. de Groot M, Hoeksma M, Blau N, Reijngoud D, van Spronsen F. Pathogenesis of cognitive dysfunction in phenylketonuria: review of hypotheses. Mol Genet Metab. [online] 2010 outubro; [acesso em 19 mar. 2019]; 99(1):S86-9. Disponível em https://www.ncbi.nlm.nih.gov/pubmed/20123477
16. Brasil. Portaria SAS/MS nº 1.307, de 22 de novembro de 2013. Protocolo Clínico e Diretrizes terapêuticas - Fenilcetonúria. Diário Oficial da União 25 nov. 2013. Seção 1; página 61
17. Hess ARB, Falcke D. Sintomas internalizantes na adolescência e as relações familiares: uma revisão sistemática da literatura. Psico-USF [online] 2013 maio-agosto; [acesso em 19 mar. 2019]; 18(2):263-7. Disponível em http://www.scielo.br/scielo.php?script=sci_arttext&pid=S1413-82712013000200010
18. Dib SA, Tschiedel B, Nery M. Diabetes melito tipo 1: da pesquisa à clínica. Arq Bras Endocrinol Metab. [online] 2008 março; [acesso em 19 mar. 2019]. 52(2):143-5. Disponível em http://www.scielo.br/scielo.php?script=sci_arttext&pid=S0004-27302008000200001
19. Siqueira MFA, Miranda OA, Bissoli LF. Neuropsychological assessment of adult pku patient on a short-term therapy with sapropterin dihydrochloride: could it be predict sapropterin-responsivess? Mol Genet Metab. [online] 2019 abril; [acesso em 12 maio 2019]. 126:328. Disponível em file:///D:/Documents/Downloads/revista%20publicada%201-s2.0-S1096719219301131-main.pdf
20. Miranda AO, Siqueira MFA, Bissoli LF. Therapeutic monitoring of sapropterin dihydrochloride responsiveness with urine organic acids chromatography analysis on filter-paper. Mol Genet Metab. [online] 2019 abril; [acesso em 12 maio 2019]. 126:316. Disponível em file:///D:/Documents/Downloads/revista%20publicada%201-s2.0-S1096719219301131-main.pdf
21. Munshi, MN. Cognitive Dysfunction in Older Adults With Diabetes: What a Clinician Needs to Know. Diabetes Care Apr 2017, 40 (4) 461-467; DOI: 10.2337/dc16-1229
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()