Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(3) - Setembro 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Síndrome de Sanfilippo IIIA: desvendando o fenótipo clínico em duas irmãs
Sanfilippo IIIA syndrome: unraveling clinical phenotypes in two sisters
Juliana Sahao1; Julia Peral1; Karolyne Cazotto1; Zumira Aparecida Carneiro1; Patricia Barros Viegas Anno2; Laura Vagnini3; Marcela Almeida1; Jacqueline Fonseca4; Debora de Cassia Tomaz2; Regina Albuquerque2; Ana Paula Andrade Hamad5; Charles Marques Lourenco1,2,3
DOI:10.31365/issn.2595-1769. v20i3p116-120
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - Ribeirão Preto - SP - Brasil
3. Centro Paulista de Diagnostico e Pesquisa, Genetica Clínica - Ribeirão Preto - SP - Brasil
4. Laboratório DLE, Genética Bioquímica - Rio de Janeiro - RJ - Brasil
5. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - Ribeirão Preto - SP - Brasil
Endereço para correspondência:
Recebido em: 12/02/2020
Aprovado em: 16/08/2020
Instituição: Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
Resumo
A mucopolissacaridose tipo III (síndrome de Sanfilippo) compreende um grupo de doenças autossômicas recessivas hereditárias, pertencentes ao grupo de doenças lisossômicas de depósito (LSD) dos mucopolissacarídeos (glicosaminoglicanos, GAGs). A doença é causada pela deficiência de uma das quatro enzimas responsáveis pela degradação do heparan sulfato, um tipo específico de GAG. Relatamos duas irmãs com diagnóstico de síndrome de Sanfilippo IIIA confirmado por testes enzimáticos e genéticos. Ainda não existe terapia específica curativa para essa enfermidade, mas o diagnóstico correto permite que as pacientes se beneficiem com tratamento sintomático e terapias de reabilitação, além de possibilitar o aconselhamento genético familiar.
Palavras-chave: Mucopolissacaridose III; Transtornos do desenvolvimento da linguagem; Proteoglicanas de heparan sulfato; Glicosaminoglicanos.
Abstract
Type III mucopolysaccharidosis (Sanfilippo syndrome) comprises a group of autosomal recessive hereditary disorders, belonging to the group of lysosomal storage diseases (LSD) of mucopolysaccharides (glycosaminoglycans, GAGs). The disease is caused by the deficiency of one of the four enzymes responsible for the degradation of heparan sulfate, a specific type of GAG. We report two sisters with Sanfilippo IIIA syndrome confirmed by enzymatic and genetic tests. Specific therapy has not yet been developed, however a correct diagnosis allows not only the benefits from symptomatic treatment and rehabilitation therapies for the patients as well genetic counselling for the families.
Keywords: Mucopolysaccharidosis III; Language development disorders; Heparan sulfate proteoglycans; Glycosaminoglycans.
INTRODUÇÃO
As doenças lisossômicas compõem um grupo de mais de 50 desordens hereditárias1 caracterizadas pela deficiência de enzimas responsáveis pela degradação de produtos metabólicos, cujo acúmulo resulta em disfunção celular, gerando, assim, as manifestações clínicas.2
As mucopolissacaridoses (conhecidas como MPSs) constituem um conjunto de patologias pertencentes ao grupo das doenças lisossomais de depósito (LSD), subdivido em pelo menos nove tipos. Entre eles, destaca-se o tipo III, também conhecido como síndrome de Sanfilippo, por ser a forma de mucopolissacaridose considerada mais comum, porém de diagnóstico mais difícil por não apresentar o fenótipo clínico típico das outras formas de MPS. A síndrome de Sanfilippo divide-se em quatro subtipos, a saber: MPS IIIA, IIIB, IIIC e IIID. Especificamente, a MPS IIIA apresenta idade de início mais precoce e, usualmente, manifestações clínicas mais graves, embora haja grande variabilidade clínica entre pacientes de diferentes famílias e mesmo dentro da própria família.1
A deficiência da enzima específica heparan N-sulfatase, responsável pela quebra de glicosaminoglicanos (GAGs), codificada pelo gene SGSH, resulta no acúmulo intralisossomal de heparan sulfato, o qual inicia um processo de neurodegeneração e neuroinflamação que, no longo prazo, desencadeia as manifestações clínicas principais da doença, acometendo primariamente o sistema nervoso central (SNC).3
Os pacientes com MPS IIIA iniciam o quadro com manifestação mental e comportamental, como dificuldade/atraso de fala e, posteriormente, hiperatividade e irritabilidade.4 Há associação com alterações fenotípicas, caracterizadas em nariz em sela, lábios cheios e polpudos, cabelos abundantes, sobrancelhas densas, pescoço curto e protrusão lingual. No exame físico, a presença de hepatoesplenomegalia também é frequente.5
RELATO DE CASO 1
Paciente, sexo feminino, 16 anos, branca, primeira filha de casal jovem e não consanguíneo, foi encaminhada para avaliação de quadro clínico de atraso de neurodesenvolvimento (particularmente da fala). Genitores referem gestação sem intercorrências; paciente nasceu de parto cesáreo, a termo, com 50 cm de comprimento, peso de 3,750 kg, APGAR 9 no 5º minuto. Não houve relato de intercorrências no período neonatal.
Marcos de neurodesenvolvimento revelaram normalidade de aquisições motoras, com estagnação da fala em torno dos dois anos de idade, quando os genitores observaram alterações comportamentais com agitação e hiperatividade crescentes. Em avaliação de rotina com pediatra, observou-se hepatoesplenomegalia com 1 ano e 3 meses, sem contudo elucidar-se a etiologia da visceromegalia na paciente. Aos 22 meses, a paciente foi submetida a adenoidectomia, por dificuldade respiratória (apresentava episódios de infecções de vias áreas de repetição). Com cerca de 5 anos de idade, fez amigdalectomia com melhora de padrão respiratório.
Em virtude do quadro clínico de estagnação de fala associado a alterações comportamentais progressivas, paciente foi avaliada em serviço de genética médica. Ao exame físico, foram observados fácies grosseira, com nariz em sela, cabelos grossos, pescoço curto, hirsutismo e lábios volumosos (figura 1), além de hepatoesplenomegalia.
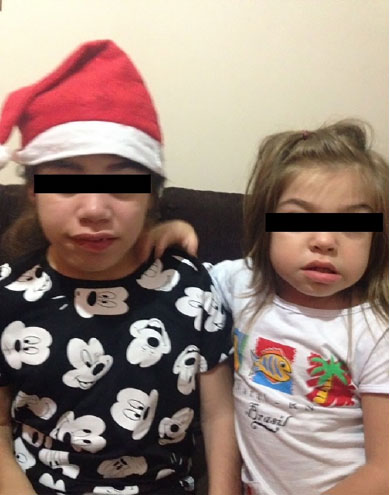
Figura 1. Pacientes 1 e 2 portadoras de MPS IIIA. Observar aspectos faciais grosseiros ("coarse facial features")
Fonte: os autores (2020).
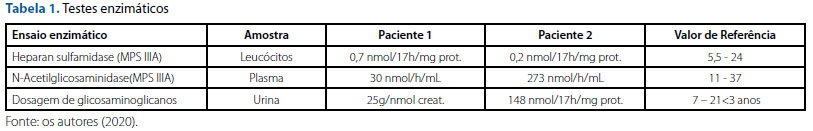
Após suspeita de doença de armazenamento lisossômico, foram solicitados testes enzimáticos sanguíneos e urinários que confirmaram o diagnóstico de síndrome de Sanfilippo IIIA (tabela 1). Posteriormente, sequenciamento genético confirmou a presença de duas mutações em heterozigose composta no gene SGSH: a deleção c.1080delC e a mutação missense c.1122C>A, ambas no exon 8 desse mesmo gene.
Paciente evoluiu com piora progressiva do quadro neurológico, com regressão completa da fala, perda de interação social (adquirindo características "autista-like") e lenta involução motora. A partir dos 10 anos, passou a apresentar maior dificuldade de deambulação, perda do controle dos esfíncteres, sendo necessário o uso de fraldas. Aos 16 anos, teve primeira crise convulsiva, caracterizada por imobilidade e rigidez por alguns segundos, seguido de sonolência. Eletroencefalograma evidenciou atividade irritativa frontotemporal à esquerda. Paciente segue em uso de anticonvulsivante para controle de crises.
De comorbidades atuais, foi diagnosticada com síndrome dos ovários policísticos e insuficiência aórtica discreta (em exame de ecocardiograma). Alimentação é feita por via oral, mas recentemente a paciente começou a apresentar engasgos frequentes. Realiza terapias de reabilitação com fonoaudiologia, fisioterapia, psicopedagogia e terapia ocupacional.
RELATO DE CASO 2
Paciente, sexo feminino, 5 anos de idade, branca, irmã da paciente 1, foi encaminhada para avaliação em virtude de a irmã ser portadora de síndrome de Sanfilippo A. Nasceu de parto cesáreo, pré-termo (36 semanas), após gestação sem intercorrência, com 49 cm de comprimento, peso de 4,650 kg, APGAR 9 no 5º minuto. Não houve relato de intercorrências no período neonatal.
Marcos motores foram considerados dentro da normalidade, mas a paciente nunca apresentou normalidade no desenvolvimento da fala, evoluindo com atraso de linguagem e obtenção de esparso vocabulário, composto de poucas palavras, nunca tendo sido capaz de juntar duas palavras em uma frase. De modo semelhante à irmã, evoluiu com alterações comportamentais, porém exibindo comportamento mais destrutivo e agressivo que irmã mais velha. De comorbidades clínicas, apresenta infecções de vias aéreas de repetição, mas não necessitou realizar qualquer tipo de intervenção cirúrgica.
Diante do quadro clínico e da história familiar positiva, paciente foi avaliada em serviço de genética, quando foram observados traços faciais grosseiros, com lábios volumosos, discreto hirsutismo corporal, ausência de visceromegalias. Testes enzimáticos (tabela 1) e genético-moleculares foram realizados, confirmando o diagnóstico de mucopolissacaridose tipo IIIA. Até o momento não apresentou sinais de crise convulsiva, segue com dificuldades respiratórias e piora de agitação comportamental e hiperatividade. Está em seguimento com terapias de reabilitação (fonoaudiologia, fisioterapia e terapia ocupacional).
DISCUSSÃO
A síndrome de Sanfilippo possui um caráter progressivo, decorrente das alterações neurodegenerativas desencadeadas a partir do acúmulo intracelular de heparan sulfato.6 Clinicamente, caracteriza-se por atraso no desenvolvimento cognitivo (principalmente da fala), mudanças de comportamento e posterior declínio de suas funções básicas, como observado nas pacientes (figura 2).
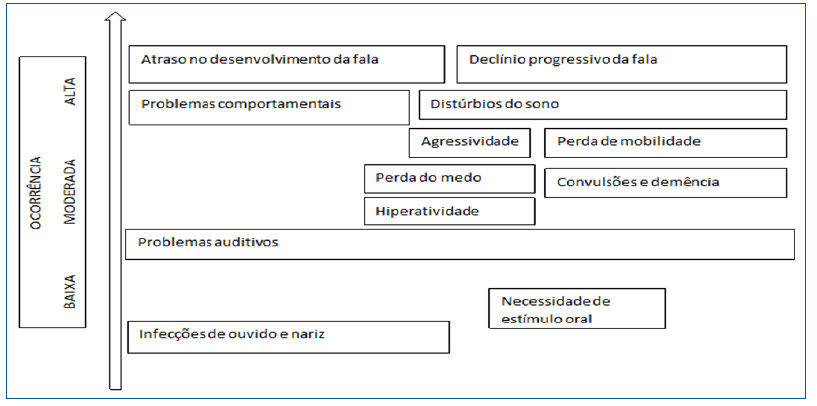
Figura 2. Progressão clínica da doença
Fonte: os autores (2020).
Alterações respiratórias são frequentes em portadores da síndrome e estão presentes nas pacientes do relato, podendo ser as primeiras manifestações da doença, ainda na ausência de alterações neurológicas relevantes.6 Facies grosseira, com nariz em sela, lábios volumosos, cabelos abundantes, sobrancelhas densas, pescoço curto e protrusão lingual apresentam-se em 76% dos casos e são também observados nas irmãs apresentadas (figura 1). Hepatomegalia, identificada na paciente 1, é observada em cerca de 67% dos pacientes e pode ser um sinal de alerta para o clínico.
Importante ressaltar que o conhecimento biológico dos médicos é essencial, para reconhecer e realizar os devidos testes enzimáticos, ou encaminhar para serviço especializado.6 Em síntese, a MPS III deve ser incluída no diagnóstico diferencial de atraso no desenvolvimento, especialmente quando associado a facies grosseira.7
As alterações de comportamento oscilam no início da doença com hiperatividade, agitação e irritabilidade/comportamento destrutivo; com a progressão da enfermidade, paciente desenvolve aparente "calmaria e tranquilidade", revelando a evolução do processo de neurodegeração. Tal situação requer dos pais ou cuidadores atenção integral e supervisão em locais de convivência coletiva com outras crianças.4 Após os 10 anos, há perda progressiva e inexorável de habilidades motoras e cognitivas2, conforme também observado na paciente 1.
A idade média de óbito na MPS IIIA ocorre na segunda década de vida, geralmente devido a complicações da progressão da doença neurológica como, por exemplo, piora da disfagia, que pode levar a episódios de broncoaspiração.8 Dessa forma, pode-se destacar a pneumonia - usualmente por broncoaspiração - como a causa mais relatada de óbito em pacientes com MPS IIIA seguido de pacientes que apresentam morte súbita sem causa identificada.9
A sobrecarga para a família devido à perda de mobilidade e autocuidado e outras habilidades prejudica sobremaneira a qualidade dos pacientes e de seus cuidadores.10 Ressaltamos a importância de se conhecer a história natural e as principais características clínicas da síndrome de Sanfilippo, notadamente pelos pediatras para reconhecimento precoce dos sinais e sintomas dessa enfermidade, visto que novas terapias para tratamento dessa doença (como terapia de reposição enzimática intratecal e terapia gênica) estão em desenvolvimento e o êxito delas dependerá da identificação de pacientes em uma fase inicial de acometimento neurológico, antes do estabelecimento de sequelas irreversíveis ao sistema nervoso central.11
CONCLUSÃO
A síndrome de Sanfilippo pode apresentar-se como uma síndrome de neurodesenvolvimento com acometimento preferencial da fala, mesmo na ausência de outros achados clínicos. Alterações comportamentais progressivas em crianças previamente hígidas podem ser os sintomas iniciais da síndrome de Sanfilippo, sendo muitos pacientes diagnosticados erroneamente como portadores de autismo.
Diante de novas possibilidades terapêuticas em desenvolvimento para essa enfermidade, é vital o reconhecimento da MPS IIIA pelos pediatras, visto que o diagnóstico precoce permite o início de medidas terapêuticas antes de sequelas irreversíveis da doença se instalarem
REFERÊNCIAS
1. Andrade F, Aldámiz‐Echevarría L, Llarena M, Couce ML. Sanfilippo syndrome: overall review. Pediatrics International. 2015 Apr 7;57(3):331-338.
2. Fedele A. Sanfilippo syndrome: causes, consequences, and treatments. Dove Press Journal. 2015 Nov 25; The Application of Clinical Genetics:269-281.
3. Wilkin J, Kerr NC, Byrd KW, Ward JC, Innaccone A. Characterization of a case of pigmentary retinopathy in Sanfilippo syndrome Type IIIA associated with compound heterozygous mutations in the SGSH gene. Ophthalmic Genetics. 2015 Sep 02;37(2):217-227.
4. Valstar MJ, Ruijter GJG, Van Diggelen OP, Poorthuis BJ, Wijburg FA. Sanfilippo syndrome: a mini-review. Journal of Inherited Metabolic Disease. 2008 Apr 04;31(2):240-252.
5. Rossier VF, Guaré R de O, Haddad AS, Ciamponi AL. Mucopolissacaridose tipo III (síndrome de Sanfi lippo): revisão e relato de casos clínicos. Rev Ibero-Am Odontopediatria 2004;7(38):326-34.
6. Buhrman D, Thakkar K, Poe M, Escolar ML. Natural history of Sanfilippo syndrome type A. Journal Of Inherited Metabolic Disease. 2013 Nov 23;37(3):431-437.
7. Kartal A. Delayed speech, hyperactivity, and coarse facies: does Sanfilippo syndrome come to mind? Journal of Pediatric Neurosciences. 2016 Jul-Sep;11(3):282-284.
8. Shapiro EG, Nestrasil I, Delaney KA, Rudser K, Kovac V, Nair N. et al. A prospective natural history study of mucopolysaccharidosis type IIIA. The Journal of Pediatrics. 2016 Jan 16;170(4):278-287.
9. Valstar MJ, Neijs S, Bruggenwirth HT, Olmer R, Ruijter GJG, Wevers RA. et al. Mucopolysaccharidosis type IIIA: clinical spectrum and genotype-phenotype correlations. Annals of Neurology. 2010 Nov 08;68(6):876-887.
10. Shapiro E, Ahmed A, Whitley C, Delaney K. Observing the advanced disease course in mucopolysaccharidosis, type IIIA; a case series. Molecular Genetics and Metabolism. 2017 Nov 28;123(2):123-126
11. Delgadillo V, O'Callaghan MdM, Gourt L, Coll MJ, Pineda M. Natural history of Sanfilippo syndrome in Spain. Orphanet Journal Of Rare Diseases. 2013 Dec 06;8(1):180-200.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()