Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(3) - Setembro 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Síndrome de Pitt-Hopkins: relato de caso com fenótipo atenuado
Pitt-Hopkins syndrome: case report of a patient with attenuated phenotype
Ana Beatriz Rodrigues da-Silva1; Caroline Yuassa1; Daniel Bussiki Santos1; Laura Lascala Cardoso1; Zumira Aparecida Carneiro1; Patricia Barros Viegas Anno2; Regina Albuquerque2; Debora de Cassia Tomaz2; Charles Marques Lourenco1
DOI:10.31365/issn.2595-1769. v20i3p111-115
1. Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirao Preto - SP - Brasil
2. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
Endereço para correspondência:
Recebido em: 06/02/2020
Aprovado em: 14/08/2020
Instituição: Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirao Preto - SP - Brasil
Resumo
A síndrome de Pitt-Hopkins (PTHS) é uma síndrome de neurodesenvolvimento rara provocada por mutações no gene TCF4, usualmente associado a atraso neuropsicomotor, crises convulsivas, distúrbios respiratórios, deficiência intelectual e dismorfias faciais. Relatamos o caso de paciente brasileira com mutação no gene TCF4 apresentando sinais clínicos compatíveis com a PTHS, embora não apresentando o característico distúrbio respiratório. A paciente apresenta uma nova mutação no gene TCF4 previamente não descrita na literatura e um fenótipo clínico mais atenuado em comparação com pacientes descritos anteriormente.
Palavras-chave: Fator de transcrição 4; Transtornos do desenvolvimento da linguagem; Epilepsia generalizada; Deficiência intelectual.
Abstract
Pitt-Hopkins syndrome (PTHS) is a rare neurodevelopmental syndrome caused by mutations in the TCF4 gene usually associated with neuropsychomotor delay, seizures, respiratory disorders, intellectual disability and facial dysmorphism. Here we report a Brazilian patient harboring mutation in TCF4 gene, showing few clinical signs of PTHS, in spite of not presenting the characteristic respiratory disorder seen in many reported patients. Our patient has a new mutation in the TCF4 gene not described in the literature so far and a more attenuated clinical phenotype compared with other patients.
Keywords: Transcription Factor 4; Language development disorders; Epilepsy, generalized;Intellectual disability.
INTRODUÇÃO
A síndrome de Pitt-Hopkins (PTHS) é um raro distúrbio de neurodesenvolvimento autossômico dominante, causado por haploinsuficiência do gene TCF4, essencial para o desenvolvimento do sistema nervoso.1,2 A haploinsuficiência do gene TCF4 pode ser causada tanto por mutação no gene quanto por deleção da região cromossômica em que esse gene está localizado (18q21.2).3 Dessa forma, não há a expressão desse gene, levando a determinados sinais e sintomas no indivíduo. Acomete homens e mulheres e possui caráter pan-étnico.4
A PTHS é caracterizada por dismorfias faciais (olhos profundos, nariz proeminente e lábios grossos com dentes espaçados), atraso global de neurodesenvolvimento (em particular da fala), presença de movimentos estereotipados e deficiência intelectual (moderada a grave).1 Quadros de epilepsia e padrões respiratórios anormais, como episódios de hiperventilação e apneia, podem estar presentes também.1 Crianças com PTHS tendem a ter uma disposição feliz, com palpitação e excitabilidade, além de características autista-like, o que leva à confusão diagnóstica com outras síndromes genéticas mais comuns de características similares, como a síndrome de Angelman e a síndrome de Rett.1
Aqui relatamos caso de síndrome de Pitt-Hopkins com fenótipo atenuado, confirmado a partir de identificação de mutação no gene TCF4.
Relato de casoPaciente do sexo feminino, 7 anos, única filha de pais não consanguíneos, foi encaminhada para avaliação de crises convulsivas associadas a atraso de fala. Antecedentes obstétricos revelaram que a genitora da paciente realizou todas as consultas pré-natais, relatou movimentação fetal a partir do sexto mês de gestação, negou intercorrências durante a gestação ou exposição a agentes teratogênicos. Referiu rotura da bolsa placentária dez dias antes do previsto, permanecendo mais de 12 horas em trabalho de parto, sendo necessário o uso de fórceps durante o parto.
Paciente nasceu a termo (39 semanas) de parto normal. Seu peso ao nascer era 2.510 gramas, com 49 centímetros (cm) de altura, 32 cm de perímetro craniano, 30 cm de perímetro torácico, 24 cm de perímetro abdominal, APGAR de 5 no 1º e 8 no 5º. Nasceu apresentando ausência de choro, cianótica e apneica, com necessidade de reanimação e uso de oxigênio.
Seus antecedentes de neurodesenvolvimento revelaram marcos motores dentro da normalidade até o primeiro ano de vida, mas após esse período observou-se atraso particularmente da fala, e que até então só falava palavras como "mãe" e "pai". Alguns dismorfismos faciais foram observados nessa época (figura 1). A partir do quarto ano de vida, tiveram início as crises tônico-clônicas. Seu histórico familiar não revelou caso semelhante ou quaisquer outras patologias de cunho genético ou hereditário.

Figura 1. Observam-se leves sinais dismórficos na paciente, especialmente em foto mais atual: frontal amplo, face triangular, queixo pontiagudo ("pointed chin"), ponta nasal bulbosa, boca grande, lábios grossos. Na primeira imagem paciente no primeiro ano de vida e, na segunda, aos sete anos de idade.
Fonte: os autores (2018).
Atualmente, consegue deambular e correr, mas não possui comunicação verbal, apenas balbucia. Possui movimentos estereotipados com as mãos ("flapping") e alterações comportamentais (hiperatividade), manifestando choro fácil, irritabilidade, agitação, ansiedade e autoagressão. Em decorrência da fisioterapia e terapia ocupacional, consegue identificar de maneira simples os indivíduos de seu convívio, apresenta riso social, porém demonstra comportamento fóbico em algumas situações.
A paciente já utilizou diversos anticonvulsivantes para tratamento (valproato de sódio, periciazina, vigabatrina, fenobarbital, lamotrigina e topiramato), sem obtenção de controle satisfatório das crises convulsivas; e também diversos medicamentos para melhorar a parte comportamental como risperidona, cloridrato de imipramina e hemifuramato de quetiapina, sem apresentar melhora dos sintomas de agitação e "autista-like".
Foi proposta a hipótese de síndrome de West a partir da presença de hipsarritmia no eletroencefalograma, padrão que não se manteve nos eletroencefalogramas subsequentes. Também já recebeu o diagnóstico de transtorno do espectro do autismo (TEA).
Considerando a complexidade do quadro da paciente envolvendo atraso de fala associado às alterações motoras e crises convulsivas, foi encaminhada a serviço de genética para investigação complementar quando realizou exames de cariótipo e CGH-Array, que não evidenciaram alterações cromossômicas; também realizou o exame de ressonância magnética, que não mostrou lesões estruturais. Diante disso e da ausência de um diagnóstico clínico estabelecido, optou-se por realizar o exame de sequenciamento exômico para acelerar o processo diagnóstico que evidenciou a presença de mutação missense em heterozigose no gene TCF4, variante c.506C>T (p.Pro169Leu), uma variante patogênica previamente não descrita na literatura, confirmando o diagnóstico de síndrome de Pitt-Hopkins na paciente.
DISCUSSÃO
A síndrome de Pitt-Hopkins (PHS) é um distúrbio raro com uma prevalência de um caso em 400.000 pessoas na população.4 Há, aproximadamente, mil casos relatados no mundo16 e muitos ainda permanecem subdiagnosticados devido à dificuldade em reconhecer a doença e sua similaridade com outras síndromes. No Brasil não há estatísticas oficiais, embora haja uma publicação prévia apresentando caso de duas pacientes gemelares afetadas simultaneamente por síndrome de Prader-Willi e síndrome de Pitt-Hopkins, além de dados da associação brasileira de síndrome de Pitt-Hopkins apontando 27 casos confirmados no país.16,17
PTHS pode ser causada tanto por mutações no gene TCF4 quanto por microdeleções cromossômicas envolvendo esse gene.3 Em geral, os casos em que ocorrem deleções envolvendo esse gene e outros genes próximos tendem a ser quadros mais graves da síndrome, podendo manifestar dismorfias mais pronunciadas e ter associação com outras malformações. Este motivo pode explicar o fato de a paciente do relato apresentar um quadro mais leve, pois trata-se de um caso envolvendo uma mutação de ponto no gene TCF4, evidenciada no exame de sequenciamento exômico.
O reconhecimento da síndrome baseia-se na suspeita clínica, cujas principais características incluem: alterações cognitivas, atraso no desenvolvimento motor e da linguagem, anormalidades respiratórias, encefalopatia epiléptica, alterações comportamentais, movimentos estereotipados e dismorfismo facial (ponte nasal alargada, narinas dilatadas, boca grande, orelhas em concha, lábio superior em forma de "arco de Cupido").5 Embora os pacientes com PTHS geralmente tenham vários sintomas sistêmicos, a maioria deles manifesta, principalmente, alterações no sistema nervoso central (SNC), quando não exclusivamente.6 Em alguns pacientes com PTHS, o envolvimento do SNC é o elemento mais marcante da doença e, muitas vezes, o único achado clínico relevante. Nossa paciente, por exemplo, não apresenta anormalidades respiratórias e não possui os aspectos dismórficos clássicos da doença, o que poderia levar ao não reconhecimento clínico de PTHS como causa do seu fenótipo neurológico.
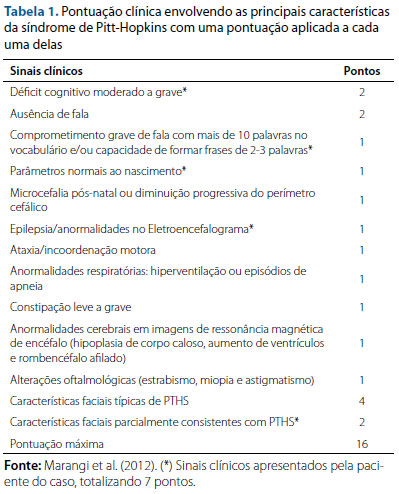
Justamente pela dificuldade de reconhecimento clínico de PTHS, desenvolveu-se um escore clínico incluindo os principais achados da síndrome, usados como critérios, com uma pontuação atribuída a cada um deles (tabela 1).7 No entanto, nossa paciente poderia não ter sido diagnosticada se fosse submetida ao escore, por conta da leve apresentação do quadro clínico e por não apresentar alguns sinais e sintomas presentes na doença com as anormalidades respiratórias, alterações na ressonância magnética, dismorfismo facial marcante, nem disfunção motora mais grave, dentre outras descritas no escore. A paciente do relato receberia pontuação de apenas 8, em um total de 16, sendo abaixo de 9 pontos considerada baixa probabilidade de ser Pitt-Hopkins para indicação dos testes genéticos específicos.
A síndrome pode cursar com características "autista-like",3 por isso muitos casos são diagnosticados com transtorno do espectro do autismo (TEA),8 como foi o caso da nossa paciente. Pacientes com PTHS têm dificuldade para interagir socialmente, movimentos estereotipados, fascínio por determinados objetos, dificuldades em mudanças de rotina, alterações comportamentais, entre outros sintomas.8 Além disso, apresenta semelhanças com outras síndromes, sobretudo síndrome de Rett (causada por mutações no gene MECP2) e de Angelman (causada por metilação anormal no locus SNRPN do cromossomo 15).9 A síndrome de Rett é uma das principais causas de atraso neurológico em meninas10 e se assemelha à síndrome de Pitt-Hopkins nos seguintes aspectos: desaceleração da velocidade de crescimento craniano; alterações respiratórias com períodos de hiperpneia intercalados por apneia; epilepsia; comportamento autista-like; movimentos estereotipados e prejuízo na habilidade de comunicação.11,12 A síndrome de Angelman também possui sobreposição clínica com PTHS e se caracteriza por retardo no desenvolvimento, ausência da fala, comportamento autista, movimentos involuntários, fácies peculiares com uma boca larga e episódios não provocados de risos e sorrisos.13
O diagnóstico de PTHS baseia-se, portanto, em achados clínicos que devem ser necessariamente confirmados por identificação de alterações no gene TCF4, incluindo sequenciamento genético-molecular (por sequenciamento Sanger ou exômico) e análise de microarray cromossômico.14 Outros exames podem ser solicitados visando identificar a presença de comorbidades em pacientes com PTHS e acompanhar a evolução clínica do paciente, como eletroencefalograma (EEG) e ressonância magnética (RNM) de encéfalo. RNM usualmente evidencia hipocampo reduzido e, algumas vezes, hipoplasia do corpo caloso e dilatação ventricular,15 mas não foram evidenciadas tais alterações em RNM da paciente do relato.
Ainda não existe uma cura para a síndrome, assim seu manejo clínico inclui terapias de suporte, tratamento sintomático e observação de comorbidades.14 Fisioterapia, terapia ocupacional e outras medidas podem auxiliar no tratamento dos pacientes.
Por fim, a raridade aparente de PTHS deve-se, provavelmente, ao subdiagnóstico, tanto pela dificuldade diagnóstica em reconhecer a síndrome devido à sua semelhança com outras doenças genéticas quanto ao não conhecimento específico de PTHS. Contudo, em razão dos avanços de sequenciamento genético, com as técnicas de sequenciamento de nova geração, está sendo possível diagnosticar pacientes com síndromes que, a princípio, não possuem todas as características clínicas da doença ou cujas características não sejam evidentes, além de identificar pacientes que poderiam não ter sido diagnosticados se fossem submetidos a escores clínicos mais tradicionais. O reconhecimento da síndrome é fundamental a fim de abordar novas perspectivas sobre a doença e estudar novas intervenções terapêuticas para garantir melhor qualidade de vida aos pacientes afetados, além de permitir o aconselhamento genético das famílias.
O QUE ESTE CASO ENSINA
Importância em colocar a síndrome de Pitt-Hopkins como diagnóstico diferencial em pacientes com transtorno do espectro autista, especialmente em pacientes com encefalopatia epiléptica de difícil controle.
Pacientes com síndrome de Pitt-Hopkins podem ter o exame CGH-Array normal, por isso há necessidade de realizar estudo molecular para exclusão ou confirmação desta hipótese diagnóstica.
A síndrome de Pitt-Hopkins também deve ser considerada como diagnóstico diferencial em pacientes com características sugestivas, porém não clássicas de síndrome de Rett e de Angelman.
Conflitos de interesseOs autores declaram a inexistência de conflitos de interesse na realização do presente trabalho.
AgradecimentosOs autores agradecem ao Prof. Dr. Charles Marques Lourenço, à Prof.ª Zumira Aparecida Carneiro e à família da paciente, pela disponibilidade e colaboração prestada.
REFERÊNCIAS
1. Amiel J, Rio M, de Pontual L, Redon R, Malan V, Boddaert N, Plouin P, Carter NP, Lyonnet S, Munnich A, Colleaux L. Mutations in TCF4, encoding a class I basic helix-loop-helix transcription factor, are responsible for Pitt-Hopkins syndrome, a severe epileptic encephalopathy associated with autonomic dysfunction. The American Journal of Human Genetics 2007; 80; 988-993.
2. Inati A, Abbas H, Korjian S, Daaboul Y, Harajeily M, Saab R. A case of Pitt-Hopkins syndrome with absence of hyperventilation. Journal of Child Neurology 2013; 28; 1698-1701.
3. Casey A, Pickard V, Ullrich C, MacNeil Z. An adapted walking intervention for a child with Pitt Hopkins syndrome. Disability and Rehabilitation: Assistive Technology 2018; 13; 25-30.
4. Fierro J, Avina D. Pitt-Hopkins syndrome: mental retardation, psychomotor and developmental delays with facial dysmorphism. Journal of Pediatric Genetics 2014; 3; 141-145.
5. Zweier C, et al. Haploinsufficiency of TCF4 causes syndromal mental retardation with intermittent hyperventilation (Pitt-Hopkins syndrome). The American Journal of American Genetics 2007; 80; 994-1001.
6. Liu Y, Guo Y, Liu P, Li F, Yang C, Song J, et al. A case of Pitt-Hopkins syndrome with de novo mutation in TCF4: clinical features and treatment for epilepsy. International Journal of Developmental Neuroscience 2018; 67; 51-54.
7. Marangi G, Ricciardi S, Orteschi D, Tenconi R, Monica M, Scarano G, et al. Proposal of a clinical score for the molecular test for Pitt-Hopkins syndrome. American Journal of Medical Genetics Part A 2012; 158; 1604-1611.
8. Balkom I, Vuijk P, Franssens M, Hoek H, Hennekam R. Development, cognition, and behaviour in Pitt-Hopkins syndrome. Developmental Medicine & Child Neurology 2012; 54; 925-931.
9. Taddeucci G, Bonuccelli A, Mantellassi I, Orsini A, Tarantino E. Pitt-Hopkins syndrome: report of a case with a TCF4 gene mutation. Italian Journal of Pediatrics 2010; 36; 12.
10. Crespo VC. Síndrome de Rett. Instituto Nacional de Pediatria 2015. Disponível em: http://repositorio.pediatria.gob.mx:8180/handle/20.500.12103/951.
11. Schwartzman JS. Síndrome de Rett. Revista Brasileira de Psiquiatria 2003; 25; 110-113.
12. Schönewolf‐Greulich B, Bisgaard A, Møller R, Dunø M, Brøndum‐Nielsen K., Kaur S, et al. Clinician's guide to genes associated with Rett‐like phenotypes: investigation of a Danish cohort and review of the literature. Clinical genetics 2017; 1-10.
13. Hong SY, Chou IC, Lin WD, Tsai FJ. A case of Pitt-Hopkins syndrome presented with Angelman-like syndromic phenotypes. BioMedicine 2016; 6; 44-46.
14. Goodspeed K, Newsom C, Morris M, Powell C, Evans P, Golla S. Pitt-Hopkins syndrome: a review of current literature, clinical approach, and 23-patient case series. Journal of Child Neurology 2018; 33; 233-244.
15. Sweetser D, Elsharkawi I, Yonker L, Steeves M, Parkin K, Thibert R. Pitt-Hopkins syndrome. GeneReviews [Internet] 2012. Disponível em: https://www.ncbi.nlm.nih.gov/books/NBK100240/.
16. Síndrome de Pitt Hopkins Brasil. [Internet] 2019. Disponível em: https://www.pitthopkins.com.br/
17. Jehee FS, de Oliveira VT, Gurgel-Giannetti J, et al. Dual molecular diagnosis contributes to atypical Prader-Willi phenotype in monozygotic twins. Am J Med Genet A. 2017;173(9):2451-2455.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()