Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(2) - Junho 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Cromossomo 13 em anel em recem nascido com raro espectro polimalformativo: relato de caso
Ring chromosome 13 in a newborn with rare polymalformative spectrum: a case report
Charbell Haddad Kury1,2,3; Isabelle Reis França Motta1; Giovanna Barreto De Oliveira Almeida1; Tathyanna Bichara De Souza Neves2; Samara Caruso Cavallaro2; Isabelly Dos Santos Silva1; Ana Lucia Batista Da-Silva1; Matheus Pessanha Paixão1; Barbara Soares de Oliveira Souza2; Leonardo Pereira Barros3
DOI:10.31365/issn.2595-1769.v20i2p79-82
1. Faculdade de Medicina de Campos, Bioquímica Médica - Campos dos Goytacazes - RJ - Brasil
2. Universidade Federal do Rio de Janeiro - Campus Macae, Pediatria - Macaé - Rio de Janeiro - Brasil
3. Hospital São João Batista Macae, Uti Neonatal - Macae - Rio de Janeiro - Brasil
Endereço para correspondência:
charbellkury@hotmail.com / charbellkury@uol.com.br
Recebido em: 10/12/2019
Aprovado em: 13/07/2020
Instituição: Faculdade de Medicina de Campos, Bioquímica Médica - Campos dos Goytacazes - RJ - Brasil
Resumo
INTRODUÇÃO: As anormalidades cromossômicas representam uma das principais etiologias dos defeitos congênitos e se apresentam por espectros variados de fenótipos, alguns com elevada morbimortalidade
OBJETIVO: descrever um caso raro de neonato com diagnóstico de cromossomo 13 em anel.
DESCRIÇÃO DO CASO: Neonato nascido de mãe primigesta, com história familiar de malformações fetais. Em rotina pré-natal, evidenciou-se holoprosencefalia lobar, hipotelorismo, comunicação interventricular ampla, genitália ambígua, quatro pododáctilos em pé esquerdo, entre outras alterações. O teste NIPT foi negativo para aneuploidias de cromossomos 13, 18, 21, X e Y. O estudo citogenético em sangue periférico por bandeamento CTG evidenciou a presença de cromossomo 13 em anel, [46,XY,r(13)] para o probando. O cariótipo dos pais foi normal.
DISCUSSÃO: As síndromes que envolvem o cromossomo 13 cursam com três fenótipos clínicos variados e o mais grave destes apresentam afecções neurológicas severas tal como a holoprosencefalia lobar bem como malformações gastrointestinais, genitália ambígua e ausência de dedos. É essencial que o aconselhamento genético surja como um importante elo para a comunicação entre os familiares e o profissional médico, visando tanto o esclarecimento e conforto da família, quanto a possibilidade de antever os cuidados necessários para o neonato
Palavras-chave:
INTRODUÇÃO: As anormalidades cromossômicas representam uma das principais etiologias dos defeitos congênitos e se apresentam por espectros variados de fenótipos, alguns com elevada morbimortalidade
OBJETIVO: descrever um caso raro de
Abstract
INTRODUCTION: Chromosomal abnormalities represent one of the main etiologies of birth defects. This condition is manifested by several phenotypes, some with high morbidity and mortality
OBJECTIVE: To describe a rare case of a newborn with 13-ring chromosome.
CASE DESCRIPTION: A newborn born to a primiparous mother with family history of fetal malformations. In prenatal routine, lobar holoprosencephaly, hypertelorism, wide interventricular communication, ambiguous genitalia, 4 left toes, among other changes were found. The NIPT test was negative for chromosome 13, 18, 21, X and Y aneuploidies. The cytogenetic study in peripheral blood by CTG banding showed the presence of ring chromosome 13, [46,XY,r(13)] for the proband
DISCUSSION: Syndromes involving chromosome 13 have three varied clinical phenotypes, the most severe of which have severe neurological disorders such as lobar holoprosencephaly as well as gastrointestinal malformations, ambiguous genitalia and absence of fingers. It is essential that genetic counseling emerges as an important link for communication between family members and the medical professional, seeking both the clarification and comfort of the family, as well as the possibility of providing the necessary care for the newborn
Keywords: Abnormal karyotype; Genetic counseling; Chromosome Aberrations.
INTRODUÇÃO
Os defeitos congênitos (DC) são alterações com origem no desenvolvimento embrionário, podendo estar presentes ao nascimento ou se manifestarem em etapas mais avançadas da vida. Em sua etiologia podem ter origem estrutural, funcional ou metabólica, sendo causadas tanto por fatores genéticos, como ambientais ou ligados às causas desconhecidas.1 Entre os DC estruturais estão as malformações congênitas (MFC) que, por sua vez ocorrem devido a uma mudança tecidual intrínseca durante o desenvolvimento normal dos tecidos/órgãos, resultando em modificações permanentes.2
As MFC representam importante problema de saúde pública, sendo responsáveis no mundo por cerca de 250.000 até um milhão de mortes anualmente.3 Estima-se que elas estejam presentes em 2 a 3% dos nascidos vivos e que possam chegar a 5% se as anormalidades cardíacas, renais e pulmonares diagnosticadas mais tardiamente forem contabilizadas.1 No Brasil, as MFC constituem a segunda causa de mortalidade infantil, determinando 11,2% dessas mortes, sendo o sistema cardiovascular o mais afetado, associado ou não a outras malformações.4,5
Os distúrbios cromossômicos são um dos grandes responsáveis pelas MFC.6 Aproximadamente 7,5% de todas as concepções apresentam anomalias cromossômicas e o aborto ocorre na maior parte dos conceptos. Devido a isso, a frequência dessas alterações entre os nascidos-vivos é inferior a 1%.6
Os humanos possuem 46 cromossomos em cada célula, sendo 22 pares de autossômicos e um par sexual (XX ou XY). O cromossomo 13 forma um dos pares. Em relação à posição do centrômero, trata-se de um cromossomo acrôcentrico, constituído de 115 milhões de pares de bases e que representa o percentual entre 3,5 a 4% do total do DNA de uma célula. As condições clínicas associadas a anormalidades estruturais deste cromossomo são: síndrome mieloproliferativa (8p11); síndrome de Feingold; tumores como retinoblastoma, leucemias e linfomas; trissomia do cromossomo 13, além de outras alterações na estrutura do cromossomo.7
A anomalia do cromossomo 13 em anel é uma alteração cromossômica estrutural rara, com incidência de 1/58,000 nascidos vivos.8 Há vários padrões de fenótipos relacionados à síndrome e isso depende do mecanismo que ocasionou sua formação. Em geral, é originada por quebra nos braços cromossômicos curtos e longos, seguidos da fusão de suas extremidades fraturadas ou por deleções, duplicações e fusão de telômeros, o que resulta em perda de material genético e, consequentemente, na geração de uma estrutura de instabilidade.9-11
Este artigo objetiva relatar o fenótipo de um recém-nascido com diagnóstico de cromossomo 13 em anel [46,XY,r(13)].
DESCRIÇÃO DE CASO
H.V.B. nasceu de parto cesariana com 37 semanas de idade gestacional (IG), pesando 1.470g, com perímetro cefálico de 39 cm e estatura de 37cm, Apgar 3 e 4, necessitando de intubação orotraqueal (TOT) na sala de parto; foi encaminhado a UTI neonatal (UTIN).
Pai, 30 anos; mãe 26 anos, GI P0 A0, casal não consanguíneo, com história familiar de malformações fetais. Pré-natal sem intercorrências infecciosas durante toda a gestação. Exame de translucência nucal (TN) com 12 semanas de IG sem alterações. Ultrassonografia obstétrica (USG) identificou imagem sugestiva de holoprosencefalia lobar. A gestante realizou o teste NIPT, que indicou baixo risco para alterações cromossômicas numéricas (aneuploidias) dos cromossomos 21, 18, 13, X, Y. Em USG morfológica realizada com 23 semanas de IG, identificaram-se holoprosencefalia lobar, hipotelorismo e defeito do septo átrio-ventricular total. Foi então solicitada avaliação por dopplerfluxometria (sem alterações). Outra USG obstétrica foi realizada, destacando-se restrição de crescimento intrauterino assimétrico (CIUR), comunicação interventricular ampla (CIV), genitália ambígua, aumento do polo encefálico com compressão do parênquima cerebral por provável hidrocefalia e quatro pododáctilos em pé esquerdo. Ecografia fetal na 28ª semana gestacional confirmou a comunicação interventricular (CIV) ampla muscular.
Admitido na UTIN sob intubação orotraqueal, o neonato foi estabilizado. O exame físico permitiu a identificação de múltiplas malformações congênitas, entre as quais distúrbio da diferenciação sexual (DDS), ectrodactilia com quatro dedos em cada mão e pé, implantação baixa das orelhas e macrocefalia com diástase hipertensa das suturas, sugestivo de hidrocefalia (Figura 1).
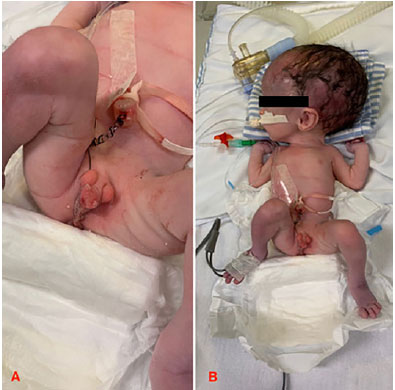
Figura 1. Foto do Probando A)Presença de distúrbio de Diferenciação sexual / Genitália ambígua. RN com cateter na região umbilical B) Ectrodactilia, com quatro dedos em cada mão e pé, implantação baixa das orelhas e macrocefalia com diástase hipertensa das suturas, sugestivo de hidrocefalia.
Durante a internação do RN, foram então realizadas tomografia de crânio, abdome e pelve, que evidenciaram redução do volume de parênquima cerebral supratentorial e tronco, ausência de estruturas da linha mediana encefálica, microftalmia e assimetria das órbitas. O cariótipo do RN foi colhido. Após cinco dias de internação, o neonato evoluiu com instabilidade hemodinâmica, quedas bruscas de saturação, bradicardia e foi a óbito. Em momento post-mortem, o estudo citogenético em sangue periférico por bandeamento CTG evidenciou a presença de cromossomo 13 em anel, [46,XY,r(13)] para o probando (Figura 2). Posteriormente, foi realizado cariótipo nos pais, com resultado normal.

Figura 2. Cariótipo do probando mostrando o cromossomo 13 em anel [46,XY,r(13)]
DISCUSSÃO
As síndromes que envolvem o cromossomo 13 cursam com quadros clínicos bastante variados. No entanto, alterações como defeitos congênitos múltiplos resultando em grave deficiência intelectual, atraso no crescimento, dismorfias craniofaciais e de membros, malformações urogenitais, cardiovasculares e neurológicas são comumente encontradas.9
As anormalidades do cromossomo 13 são divididas em três diferentes grupos de acordo com a região em que ocorre a deleção neste cromossomo. O primeiro grupo relaciona-se à deleção do conteúdo cromossômico mais proximal à banda 13q32, cursando com fenótipo de deficiência intelectual leve ou moderado, bem como de crescimento. Por sua vez, o segundo grupo refere-se a uma deleção mais distal com ligação das extremidades e inclui a região 13q32, amplificando-se as alterações, que comumente têm como fenótipo a microcefalia, hidrocefalia, ausência de polegares, genitália ambígua, malformações gastrointestinais bem como o achado deste relato de holoprosencefalia lobar (malformação cerebral complexa causada pela clivagem incompleta do prosencéfalo). Já o terceiro grupo associa-se à interrupção em 13q33-q34 e está frequentemente relacionado com o fenótipo de deficiência intelectual grave, microcefalia, hipertelorismo verdadeiro e presença de ponte nasal baixa ou plana.7,10,13 O caso descrito apresenta diversas alterações congênitas maiores, sugerindo que o mesmo se encontra no segundo grupo da classificação.
Outro dado importante trata do teste NIPT negativo no paciente relatado. Trata-se de um exame não invasivo indicado para detectar aneuploidias e algumas microdeleções cromossômicas, tal como a microdeleção 22q11.2, relacionada à síndrome de DiGeorge. Entretanto, sabe-se que na presença de malformações congênitas complexas como holoprosencefalia, o NIPT não está indicado devido à possibilidade de existirem outras aberrações cromossômicas, assim como a encontrada neste estudo, preferindo-se a realização de exames genéticos mais específicos obtidas por amniocentese.14
Um outro aspecto a ser debatido é a taxa de mortalidade em pacientes com malformações congênitas. Estudo australiano de coorte que avaliou a presença de defeitos congênitos em bebês acima de 20 semanas e 400g em um período de 2004 e 2009, evidenciando taxa de mortalidade de 7,2% nos primeiros cinco anos de vida.15 Entretanto, quando se fracionaram os dados para o tipo de fenótipo clínico encontrado, verificou-se que dos cinco casos de holoprosencefalia estudados, houve 100% de taxa de óbitos.15
Frente a este caso, os pais do probando foram encaminhados ao aconselhamento genético com profissional especialista. Essa prática permite avaliar a presença de possíveis anormalidades cromossômicas estruturais dos pais, sabendo-se que mesmo se um dos genitores tivesse anormalidades cromossômicas estruturais balanceadas, estas não formariam fenótipo, mas poderiam gerar gametas desbalanceados. O aconselhamento genético também possibilita que, frente a um diagnóstico, a família e/ou paciente recebam as informações e apoio emocional para lidar com determinada doença, tomar decisões e acessar questões apropriadas.10,12,16
É fato que o conhecimento antenatal das malformações possibilitado por um bom acompanhamento pré-natal auxiliou positivamente nesse caso, antevendo os cuidados necessários e indispensáveis para o neonato. Vale ressaltar a extrema importância do papel do profissional médico que, desde o início da investigação diagnóstica, orientou a família sobre os possíveis desfechos e cuidados de saúde necessários para propiciar melhor qualidade de vida para a criança, permitindo, assim, além da promoção de saúde para esse paciente, a possibilidade de acompanhamento psicológico minucioso para os progenitores antes e depois do nascimento da criança, valorizando-se os processos de sofrimento e luto com empatia e respeito às questões religiosas e à decisão da família.
O caso em tela ressalta a importância de os profissionais estarem atentos ao diagnóstico acurado das MFC, em especial desde a gestação através do atendimento pré-natal, não somente pelo obstetra, mas também pelo pediatra. Desta forma, pode-se assegurar a construção do vínculo e o adequado aconselhamento e suporte pós-natal à família.
REFERÊNCIAS
1. Silva JHD, Terças ACP, Pinheiro LCB, França GVA, Atanaka M, Schüler-Faccini L. Profile of congenital anomalies among live births in the municipality of Tangará da Serra, Mato Grosso, Brazil, 2006-2016. Epidemiol Serv Saude. 2018; 27(3):e2018008. Doi: 10.5123/S1679-49742018000300017. PMID: 30365695.
2. Mendes IC, Jesuino RSA, Pinheiro DS, Rebelo ACS. Anomalias congênitas e suas principais causas evitáveis: uma revisão. Rev Med Minas Gerais. 2018; 28:e-1977. Doi: http://dx.doi.org/10.5935/2238-3182.20180011
3. Liu L, Oza S, Hogan D, Perin J, Rudan I, Lawn JE, Cousens S, Mathers C, Black RE. Global, regional, and national causes of child mortality in 2000-13, with projections to inform post-2015 priorities: an updated systematic analysis. Lancet. 2015 Jan 31; 385(9966):430-40. doi: 10.1016/S0140-6736(14)61698-6. Erratum in: Lancet. 2016 Jun 18;387(10037):2506. PMID: 25280870.
4. Horovitz DD, Llerena JC Jr, Mattos RA. Atenção aos defeitos congênitos no Brasil: panorama atual. Cad Saude Publica. 2005; 21(4):1055-64. Doi: 10.1590/s0102-311x2005000400008. Epub 2005 Jul 11. PMID: 16021243.
5. Leite DL, Miziara H, Veloso M. Malformações cardíacas congênitas em necropsias pediátricas: características, associações e prevalência. Arq. Bras. Cardiol. 2010; 94(3):294-299. DOI: http://dx.doi.org/10.1590/S0066-782X2010000300003
6. Pereira TM, Oliveira ARCP, Teixeira ACZ, Jesus AN, Rodrugues MG, Agostinho MAB, et al. Frequência das Anormalidades Cromossômicas: Importância para o diagnóstico citogenético. Arq Ciênc Saúde. 2009; 16(1):31-33w
7. National Institutes of Health - NIH. Department of Health & Human Services. U.S. National Library. Lister Hill National Center for Biomedical Communications. Genetics Home Reference: Chromossome 13. Access: May 15, 2020. https://ghr.nlm.nih.gov/chromosome/13#conditions
8. Liao C, Fu F, Zhang L. Ring chromosome 13 syndrome characterized by high resolution array based comparative genomic hybridization in patient with 47, XYY syndrome: a case report. J Med Case Rep. 2011; 5:99. Doi: 10.1186/1752-1947-5-99. PMID: 21396087; PMCID: PMC3063811.
9. Guilherme, RS. Estudo clínico e citogenético-molecular de pacientes portadores de cromossomos autossômicos em anel [master's thesis]. São Paulo (SP): Unifesp; 2010. Disponível em: http://repositorio.unifesp.br/handle/11600/9498
10. Silva-Grecco RL, Palhares HMC, Lopes VLGS, Balarin MAS. Quadro polimalformativo com cariótipo 46,XYr (13): eelato de caso. Rev. Med Minas Gerais. 2006; 16(4):216-218.
11. Rodriguez, J M A. Aberraciones cromossómicas em um hospital pediátrico de tecer nível. Anillos de los cromosomas 13 y 18. Revista Iberoamericana de las Ciencias de la Salud. 2013; 3(2).
12. Ziot, R. Anomalias congênitas em natimortos e neomortos: o papel do aconselhamento genético [master´s thesis]. Rio de Janeiro: Fiocruz; 2008. Disponível em: https://bvssp.icict.fiocruz.br/pdf/ZlotRenata.pdf
13. Tratado de Pediatria: Sociedade Brasileira de Pediatria. In: DAR Burns..., et al. 4. ed. Barueri-SP: Manole; 2017.
14. Da Fonseca EB, Cruz J, Sá RA, Renzo JCD, Nicolaides K. Screening for aneuploidies in the first trimester of pregnancy: evolution from maternal age to cell-free DNA testing in maternal blood. Femina. 2014; 42(2).
15. Schneuer FJ, Bell JC, Shand AW, Walker K, Badawi N, Nassar N. Five-year survival of infants with major congenital anomalies: a registry based study. Acta Paediatr. 2019; 108(11):2008-2018. Doi: 10.1111/apa.14833. Epub 2019 Jun 13. PMID: 31046172.
16. Brunoni D. Aconselhamento genético. Ciênc. saúde coletiva. 2002; 7(1):101-107. Doi: http://dx.doi.org/10.1590/S1413-81232002000100009.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()