Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(2) - Junho 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Doença de pompe em pediatria: o caráter multifacetado de uma não tão rara doença de armazenamento lisossômico
Pompe disease in pediatrics: the multiple faces of a not so rare lysosomal storage disorder
João Pedro Camargo Campanholi1; Jonas de-Freitas1; José Paulo Aldério Almeida1; Luiz Henrique Abbes Guerra1; Marcela Almeida1; Laura Vagnini2; Jacqueline Fonseca3; Regina Albuquerque4; Zumira Aparecida Carneiro1; Fernando Silva Ramalho5; Charles Marques Lourenco1,2,4
DOI:10.31365/issn.2595-1769.v20i2p72-78
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Centro Paulista de Diagnóstico e Pesquisa, Genética Clínica - Ribeirão Preto - SP - Brasil
3. Laboratório DLE, Bioquímica Genética - Rio de Janeiro - RJ - Brasil
4. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
5. Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Patologia e Medicina Legal - Ribeirão Preto - SP - Brasil
Endereço para correspondência:
Recebido em: 04/04/2020
Aprovado em: 16/04/2020
Instituição: Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
Resumo
INTRODUÇÃO: A doença de Pompe ou doença de depósito de glicogênio tipo II é uma rara enfermidade caracterizada pela deficiência da enzima alfa glicosidase ácida (GAA), resultando em acúmulo de glicogênio nos tecidos, principalmente músculo cardíaco, esquelético e liso.
RELATO DE CASOS: CASO 1 - Paciente masculino, um mês de idade, filho de casal não-consanguíneo, encaminhado para investigação de cardiomiopatia e hepatomegalia. Apresentava aumento de transaminases, além de leve elevação de CPK e CK-MB. Nas primeiras avaliações, não apresentava sinais de hipotonia muscular, evoluindo posteriormente com sinais progressivos de hipotonia e disfunção cardíaca. Diante disso, realizaram-se ensaios enzimáticos para a deficiência de alfa-glicosidase, compatíveis com a doença de Pompe. Terapia de reposição enzimática (TRE) foi iniciada no quarto mês de vida, mas a criança evoluiu com choque cardiogênico e séptico, e falecendo.
CASO 2 - Paciente feminino, 13 anos, filha de pais não consanguíneos, encaminhada para avaliação de quadro de escoliose progressivo, com sinais de fraqueza muscular (predominantemente no pescoço) e sintomas respiratórios associados. Inicialmente houve suspeita de doença do tecido conectivo, mas após extensa investigação, confirma-se diagnóstico de forma tardia de doença de Pompe após teste enzimático e estudo molecular do gene GAA. Inicia TRE aos 16 anos, evoluindo com relativa estabilidade do quadro clínico.
CONCLUSÃO: A doença de Pompe compreende amplo espectro de manifestações, dificultando assim a detecção clínica, visto que é confundida com outras enfermidades mais comuns. Por ser uma doença atualmente passível de tratamento, diagnóstico precoce é fundamental para melhor prognóstico.
Palavras-chave: Doença de depósito de glicogênio tipo II; Doenças por armazenamento dos lisossomos; Hipotonia muscular; Escoliose; Terapia de reposição de enzimas.
Abstract
INTRODUCTION: Pompe disease or glycogen storage disease type II is characterized by the deficiency of the enzyme acid alpha-glucosidase (GAA), resulting in an accumulation of glycogen in tissues, mainly cardiac, skeletal and smooth muscle.
CASE REPORT: CASE 1 - Male patient, 1 month old, born to a non-consanguineous couple, referred for investigation of cardiomyopathy and hepatomegaly, with an increase in transaminases, in addition to a slight increase in of CPK and CK-MB. In the first evaluations, the patient showed no signs of hypotonia. The child showed progressive signs of hypotonia and cardiac dysfunction. Thus, enzymatic assays were carried out for the alpha-glucosidase deficiency, compatible with Pompe disease. Enzyme replacement therapy (ERT) was started in the fourth month of life, but the child evolved with cardiogenic shock and septic, and died.
CASE 2 - Female patient, 13 years old, born to non-consanguineous parents, presented with progressive scoliosis, signs of muscle weakness (predominantly in the neck) and respiratory symptoms associated. After extensive investigation, late-onset Pompe disease is diagnosed, after enzymatic testing and GAA molecular analysis. She started ERT at 16 years, resulting in relatively stability of her clinical picture.
CONCLUSION: Pompe disease comprises a wide spectrum of manifestations, making clinical detection difficult, since it can be misdiagnosed with other more common diseases. Since it is currently a treatable disorder, early diagnosis is critical for better prognosis.
Keywords: Glycogen storage disease type II; Lysosomal storage diseases; Muscle hypotonia; Scoliosis; Enzyme replacement therapy.
INTRODUÇÃO
A doença de Pompe ou doença de depósito de glicogênio tipo II (OMIM 232.300) é uma rara enfermidade autossômica recessiva, caracterizada pela deficiência da enzima alfa glicosidase ácida (GAA),1 também chamada de maltase ácida, uma hidrolase responsável pela degradação do glicogênio lisossomal.2 Foi descrita pela primeira vez em 1932 por Joannes Cassianus Pompe, em uma menina de sete meses que morreu de cardiomiopatia.3,4 Em 1954, foi classificada como doença de depósito de glicogênio tipo II e, em 1963, tornou-se o primeiro distúrbio de armazenamento lisossomal identificado.3,4
A deficiência da alfa glicosidase ácida leva a um acúmulo de glicogênio nos tecidos (figura 1), principalmente na musculatura cardíaca, esquelética e lisa, enquanto outros tecidos podem ser afetados com menor frequência.3,4
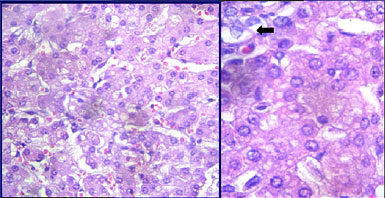
Figura 1: Aumento maciço de glicogênio intracelular (seta escura).
Fonte: os autores (2019).
A doença de Pompe pode ocorrer em qualquer idade e apresenta notável variação fenotípica, bem como diferentes graus de progressão e acometimento dos órgãos envolvidos. Classicamente, é dividida em forma infantil e forma tardia (juvenil e adulta).5
A forma infantil (doença de Pompe "clássica"), apresenta-se com cardiomiopatia, resultado do armazenamento excessivo de glicogênio no coração. Cardiomegalia maciça é evidente na radiografia de tórax e a ecocardiografia fornece evidência de aumento da espessura das paredes ventriculares e do septo interventricular.6 O eletrocardiograma geralmente mostra intervalos PR curtos e complexos QRS elevados; a síndrome de Wolf-Parkinson-White tem sido relatada em alguns pacientes. Fraqueza muscular e insuficiência respiratória progressivas são as outras características clínicas fundamentais.7,8 Os pacientes comumente apresentam organomegalia (hepatomegalia, esplenomegalia, macroglossia) e dificuldades de alimentação. Essa forma da doença geralmente culmina na morte no primeiro ano de vida devido à insuficiência cardiorrespiratória.
Alguns pacientes têm uma forma da variante infantil (doença de Pompe infantil "não clássica") com início dentro dos seis primeiros meses de idade, caracterizada por sintomas musculares e respiratórios precoces, porém sem apresentar cardiomiopatia, o que impacta em maior sobrevida em comparação com a forma infantil clássica.9
Na forma juvenil, o quadro clínico é caracterizado por miopatia axial e proximal, com envolvimento cardíaco ausente ou leve e morte antes do final da terceira década de vida. A forma adulta apresenta miopatia proximal progressiva, além de fraqueza muscular axial, desconforto respiratório e, geralmente, não há envolvimento cardíaco. Recentemente, arteriopatia dilatada, dolicoectasia da artéria basilar, hemorragias intraparenquimatosas e micro-hemorragias também foram relatadas em pacientes com doença de Pompe de início tardio.10
Relatamos aqui, a partir de dois casos clínicos, o espectro de manifestações dessa doença e a respectiva evolução.
RELATO DE CASOS
Caso
Paciente do sexo masculino, um mês de idade, filho de um jovem casal, saudável e não consanguíneo, foi encaminhado para investigação por apresentar cardiomiopatia e hepatomegalia. Inicialmente observou-se hepatomegalia em consulta de pediatria de rotina. Não houve queixa de dispneia ou dificuldade de sucção. Em avaliações subsequentes, ao se realizar exame radiográfico, observou-se aumento da área cardíaca, confirmando-se, posteriormente, a cardiomegalia pelo ecocardiograma; pelo ultrassom abdominal, confirmou-se a hepatomegalia. Exames laboratoriais revelaram aumento de transaminases que girava em torno de 250 até 400 U/L, além de leve elevação de CPK (413 , do valor de referência < 195) e elevação de CK-MB (54, do valor de referência < 25).
Nas primeiras avaliações, paciente não apresentava sinais de hipotonia muscular. Foram solicitados testes bioquímicos gerais e uma triagem geral para erros inatos do metabolismo, a qual mostrou positividade para o teste de azul de toluidina. Em vista disso, a investigação foi direcionada para mucopolissacaridoses (MPSs), visto que o azul de toluidina é um teste de triagem para essas doenças e também pelo fato de o paciente apresentar cardiomiopatia hipertrófica. Enquanto se investigavam MPSs, a criança mostrou sinais progressivos de hipotonia a partir do 3º mês de vida (com perda de controle cefálico e maior dificuldade para levantar os braços e segurar objetos) e a função cardíaca (fração de ejeção) piorou.
Diante disso, mesmo sem os resultados finais da investigação para MPSs, ensaios enzimáticos para a deficiência de alfa-glicosidase foram feitos em papel filtro; resultados preliminares foram compatíveis com a doença de Pompe e, mais tarde, ensaio enzimático foi feito em leucócitos e fibroblastos para confirmação de diagnóstico. Sequenciamento do gene GAA evidenciou que paciente era heterozigótico para duas mutações: c.2481+102_2646+31del (p.Gly828_Asn882del) e c.2560C>T (p.Arg854X).
TRE (Recombinante GAA , rGAA, MyozymeT) foi iniciada no quarto mês de vida, mas a criança já apresentava graves complicações cardíacas e, antes da 3ª infusão, desenvolveu choque cardiogênico e séptico, causando sua morte. Resultados da testagem do estado de imunidade cruzada (CRIM) foram finalizados logo após óbito do paciente e revelaram status CRIM-negativo. Avaliação post-mortem confirmou achados de maciça cardiomegalia (figura 2).
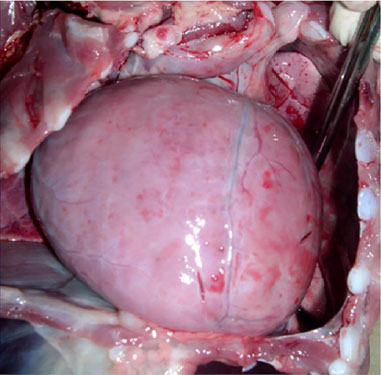
Figura 2: Presença de cardiomegalia em exame de necrópsia do paciente.
Fonte: os autores (2019).
Caso 2
Paciente do sexo feminino, 23 anos, filha de pais não consaguíneos e jovens à época da sua concepção, foi encaminhada para avaliação de escoliose "idiopática" progressiva (figura 3). Sem intercorrências relatadas durante gestação, paciente nasceu a termo, de parto normal, com peso em torno de 3kg, obtendo alta no 2º dia de vida. Neurodesenvolvimento dentro das normalidade.
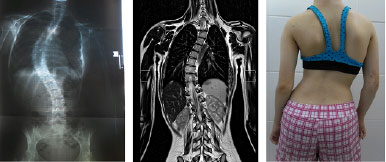
Figura 3. Escoliose de grau moderado a grave.
Fonte: os autores (2019).
Aos 13 anos, iniciou-se um quadro de escoliose, de caráter progressivo, associado a sinais de fraqueza muscular, predominantemente no pescoço e sintomas respiratórios. A princípio, foi consultada por alguns ortopedistas e fisioterapeutas para investigação de quadro de escoliose progressivo associado à frouxidão ligamentar, que a encaminharam para avaliação genética por suspeita de uma doença do tecido conectivo geneticamente determinada.
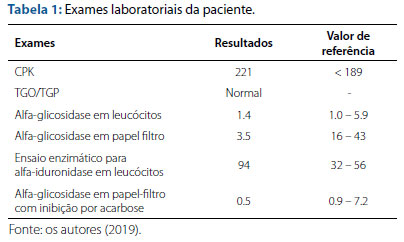
A primeira consulta com a genética foi realizada aos 14 anos de idade e, na suspeita de Pompe, foram solicitados vários exames, como: biópsia de pele e músculo, ressonância de coluna cervical e torácica, ressonância de músculos de coxa direita e esquerda e de músculos de perna direita e esquerda, raio-X, espirometria, eletrocardiograma, ecocardiograma. Os resultados dos exames foram demonstrados nas tabelas 1 e 2. Além desses, foi feito também estudo molecular do gene GAA, que evidenciou presença de mutação heterozigota c.-32-13T>G associada a outras variantes de significado clínico incerto.
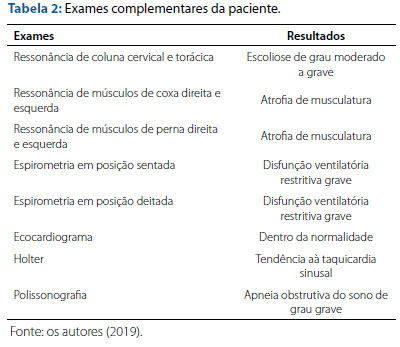
Após avaliação de tais resultados, foi diagnosticada doença de Pompe e solicitado tratamento com TRE. Aos 16 anos, conseguiu a liberação do medicamento e, por conseguinte, foi iniciada a TRE e desde então iniciou acompanhamento com fisioterapia e nutricionista.
Após dois anos de início da TRE e diante do quadro de escoliose importante que a paciente já apresentava, optou-se em abordagem cirúrgica. Realizaram, ao todo, três abordagens cirúrgicas para correção da escoliose. A primeira cirurgia consistiu na colocação de uma haste para correção da escoliose; paciente evoluiu, porém com complicação no pós-operatório, em virtude de compressão medular com perda de sensibilidade em membros inferiores. Consequentemente, realizou-se nova abordagem para retirada da haste e descompressão medular. Paciente, contudo, evoluiu com parada cardiorrespiratória no pós-operatório, sendo encaminhada para CTI, onde permanecu internada por duas semanas para recuperação clínica. Por fim, foi submetida a nova cirurgia para recolocação da haste após obter alta da CTI, evoluindo sem complicações no pós-operatório.
Atualmente, apresenta escoliose de 30° e faz acompanhamento regular com fisioterapia (duas sessões de fisioterapia pulmonar e uma de fisioterapia motora por semana) e natação dois dias na semana. Repete exames de seis em seis meses, sendo avaliada anualmente pela ortopedia, neurologia e pneumologia para acompanhamento da doença e monitorização de possível progressão dos sintomas.
DISCUSSÃO
A doença de Pompe (também conhecida como doença de depósito do glicogênio tipo II ou glicogenose tipo II) é uma doença rara, subdiagnosticada, com incidência total estimada em 1:40.000 nascidos vivos, pertencente ao grupo conhecido como doenças de depósito lisossômico (DDL).11,12,13 Caracteriza-se pelo acúmulo de glicogênio nos lisossomos, decorrente de alterações na atividade (baixa ou anormal) da enzima alfa-glicosidase ácida (figura 4).
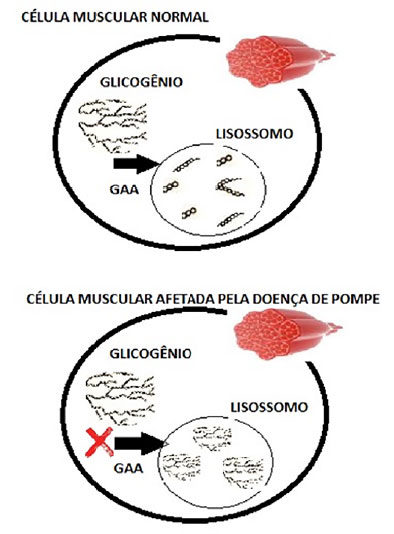
Figura 4: Deficiência de alfa-glicosidase ácida (GAA) e acúmulo de glicogênio nos lisossomos.
Esta enzima participa do processo de degradação do glicogênio em glicose dentro das fibras musculares, sendo que mutações variadas no gene da GAA induzem a diferentes graus da deficiência da enzima, podendo levar a deficiência parcial ou completa.14 O acúmulo de glicogênio lisossômico pode ocorrer em diferentes tecidos, sendo as células da musculatura lisa, esquelética e cardíaca as mais afetadas.15
O espectro clínico da doença de Pompe tem uma grande variação, o que pôde ser observado nos casos apresentados, com significativas diferenças relacionadas à idade do início dos sintomas , progressão da doença e ao fenótipo clínico, o que torna o diagnóstico precoce difícil.16,17
Na forma infantil, em que a atividade GAA está quase ou totalmente ausente, como o ocorrido com o paciente 1, a doença se manifesta comumente por cardiomiopatia hipertrófica progressiva, hipotonia (que pode anteceder a cardiomiopatia ou se apresentar posteriormente a ela, como também foi evidenciado no mesmo paciente, visto que, no início do quadro, não apresentava ainda sinais de hipotonia), dificuldade respiratória e atraso no desenvolvimento motor. Usualmente, ocorre progressão para insuficiência cardíaca e respiratória durante o primeiro ano de vida, o que tende a ser fatal, levando ao óbito antes dos dois anos de vida.18
Pacientes com doença de Pompe de início tardio, contudo, apresentam comprometimento muscular em graus variados (conforme observado na paciente 2) e podem evoluir com insuficiência respiratória decorrente do envolvimento diafragmático, levando à dependência de suporte ventilatório ou a óbito na ausência de terapia específica.11,19
Na avaliação de um paciente suspeito de ter Pompe, o teste laboratorial deve conter creatina quinase (CPK) sérica, transaminase glutâmico-oxalacética (TGO), transaminase glutâmico-pirúvica (TGP), lactato desidrogenase (LDH) e tetrassacarídeos em sangue e urina. Vários testes podem ser usados para demonstrar a diminuição da atividade enzimática da GAA e/ou a mutação do gene responsável que pode originá-la.7 A pesquisa da atividade enzimática na gota de sangue seco em papel-filtro atualmente vem sendo o primeiro teste diagnóstico a ser usado para investigação por conta de sua acessibilidade e disponibilidade, o que foi essencial para levantar a suspeita de doença de Pompe na paciente 2 e redirecionar a investigação do paciente 1.
O resultado deve ser confirmado em um segundo ensaio em outro tecido, como em linfócitos, leucócitos, fibroblastos ou tecido muscular; porém, mais modernamente, vem-se optando em realizar sequenciamento direto do gene GAA ao se encontrar deficiência enzimática na gota de sangue seco em papel-filtro.20 A presença de mutações bialélicas no gene GAA confirma o diagnóstico bioquímico, sem necessidade de testes enzimáticos adicionais ou realização de exames mais invasivos como biópsia de músculo.20,21
O diagnóstico a partir do ensaio enzimático em fibroblastos (padrão-ouro) pode fornecer, em algumas situações, informações sobre o prognóstico da doença a partir da atividade enzimática residual encontrada, porém nem sempre essas correlações são possíveis, em particular nos casos de Pompe de início tardio. No caso de um resultado inconclusivo ou negativo, persistindo a hipótese de doença de Pompe, é necessário realizar uma biópsia muscular para investigação, com a ressalva de que as formas mais tardias da doença podem apresentar distribuição não homogênea do depósito de glicogênio, resultando também em falsos-negativo.20,21
Em se tratando de uma doença geneticamente determinada, o tratamento deve ser entendido como um conjunto de medidas para curar ou diminuir o impacto da doença. Na doença de Pompe incluem-se, por um lado, o controle dos sintomas e das complicações derivadas da doença, e por outro, a implementação de uma terapia específica que tenta substituir a enzima ausente ou deficiente (TRE). O tratamento de reposição enzimática foi testado pela primeira vez em 1973 e tornou-se disponível nos anos 90, revolucionando o tratamento da doença.4
O diagnóstico de doença de Pompe de início infantil geralmente é feito em torno de 3-6 meses, sendo que apenas aproximadamente 8% das crianças sobrevivem além de um ano de vida sem tratamento.22 Em 2006, a terapia de reposição enzimática (TRE) foi aprovada para tratamento da doença de Pompe. Para proporcionar melhores resultados, o tratamento com TRE deve ser realizado o mais precoce possível, evitando danos musculares irreversíveis,23 o que nem sempre é possível, conforme observado no paciente 1. Embora este tenha sido encaminhado precocemente para investigação, a suspeita de doença de Pompe foi feita quando paciente já estava em evidente deterioração cardíaca e respiratória. Nesse contexto, a TRE não se mostrou capaz de impedir a progressão para o óbito, pois ja havia diversas sequelas irreversíveis da enfermidade.
Dada a gravidade da forma infantil clássica da doença de Pompe, alguns estados americanos colocaram essa doença como parte do teste de triagem neonatal, visto que é possível pesquisar a atividade enzimática GAA no papel filtro para se identificar precocemente essa forma de doença de Pompe em uma fase assintomática.24 Taiwan foi o pioneiro no programa de triagem neonatal para a doença de Pompe, obtendo sucesso desde 2005.25,31 Comparando-se os riscos e benefícios da TRE precoce em pacientes com suspeita de doença de Pompe de início infantil, os resultados mostraram-se eficazes em relação aos efeitos adversos.26 Consequentemente, quando o tratamento é realizado da forma mais rápida possível, melhores resultados clínicos são obtidos.
Em relação à paciente de início tardio, a apresentação de "escoliose idiopática" como manifestação de doença neuromuscular não é algo tão raro. De acordo com o Pompe Registry,27 há relação entre o impacto da escoliose em pacientes com doença de Pompe, pois com o acúmulo de glicogênio nos tecidos, há acometimento da musculatura esquelética axial, facilitando a formação de escoliose na coluna toraco-lombar.28
Ademais, outros estudos mostram que, em pacientes com doença de Pompe avaliados através da idade, a escoliose foi mais prevalente em crianças e jovens.29 Outro dado importante observado foi que pacientes com escoliose possuem função respiratória pior do que aqueles sem escoliose, o que pôde ser observado na paciente 2, que apresentava alteração expressiva da capacidade respiratória. Dessa forma, os médicos que se depararem com pacientes com problemas neuromusculares, apresentando fraqueza e escoliose, devem considerar a possiblidade de diagnóstico diferencial com a doença de Pompe.24,30,32
Em face do caráter multifacetado de apresentação dessa enfermidade tanto em pacientes pediátricos quanto pacientes adultos, foram elaborados diversos critérios de suspeita diagnóstica em diferentes guias clínicos de países distintos.32,33 Apesar de pequenas diferenças entre eles, todos apontam para a necessidade de se realizar o diagnóstico mais precoce possível do paciente para que o manejo adequado seja oferecido.,17,18
O seguimento de pacientes afetados por essa enfermidade deve envolver equipes interdisciplinares para abordar os diferentes problemas clínicos.20 A equipe de trabalho deve ser integrada por médicos (neurologista, clínico, pneumologista, cardiologista, nutricionista, gastroenterologista), cinesiologistas, terapeutas físicos, ocupacionais e de linguagem.17
Em suma, a doença de Pompe constitui um desafio diagnóstico devido ao seu caráter multifacetado, afetando desde bebês até adultos, com expressão clínica variável, abrangendo sintomas variados, como cardiomiopatia hipertrófica, hipotonia, insuficiência diafragmática, fraqueza muscular axial e escoliose progressiva. Em se tratando de uma doença agora passível de tratamento, o diagnóstico precoce torna-se essencial para que os pacientes possam receber a terapêutica adequada antes de sequelas irreversíveis (e complicações fatais) ocorram.
REFERÊNCIAS
1. Güngör D, et al. Survival and associated factors in 268 adults with Pompe disease prior to treatment with enzyme replacement therapy. Orphanet journal of rare diseases. 2011; 6(1):34.
2. Hagemans MLC, et al. Clinical manifestation and natural course of late-onset Pompe's disease in 54 Dutch patients. Brain. 2005; 128(3):671-677.
3. Kishnani PS, et al. Pompe disease diagnosis and management guideline. Genet Med. 2006; 8(5): 26788.
4. Cupler EJ, et al. "Consensus treatment recommendations for late-onset Pompe disease" Muscle & nerve. 2011; 45(3):319-33.
5. Brito-Avô L, et al. Diagnosis recommendations for late-onset Pompe disease. Acta medica portuguesa. 2014; 27(4): 525-529.
6. Martínez M, et al. Infantile-onset Pompe disease with neonatal debut: A case report and literature review. Medicine. 2017; 96(51).
7. Di Rocco M, Buzzi D, Taro M. Glycogen storage disease type II: clinical overview. Acta myologica. 2007; 26(1):42.
8. Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: literature review and case series. Neurol Clin. 2014;32(3):751-ix. doi:10.1016/j.ncl.2014.04.010
9. Teener JW. Late-onset Pompe's disease. In: Seminars in neurology. Thieme Medical Publishers; 2012. p. 506-511.
10. Laforet P, et al. Dilative arteriopathy and basilar artery dolichoectasia complicating late-onset Pompe disease. Neurology, v. 70, n. 22, p. 2063-2066, 2008.
11. Kishnani PS, Howell RR. Pompe disease in infants and children. J. Pediatr. 2004; 144(5):35-43.
12. Martiniuk F, et al. Carrier Frequency for Glycogen Storage Disease Type II in New York and Estimates of Affected Individuals Born with the Disease. American Journal of Medical Genetics. 1998; 79:69-72.
13. Sixel BS, et al. Manifestações respiratórias na doença de Pompe de início tardio: Uma série de casos no Brasil. JBP. 2017; 43(1):54-59.
14. Bembi B, et al. Management and treatment of glycogenosis type II. Neurology. 2008; 71(23-supl 2):12-36.
15. Savegnago AK, et al. Revisão sistemática das escalas utilizadas para avaliação funcional na doença de Pompe. Rev Paul Pediatr. 2012; 30(2): 272-277.
16. Boentert M, et al. Practical Recommendations for Diagnosis and Management of Respiratory Muscle Weakness in Late-Onset Pompe Disease. International Journal of Molecular Sciences; 2016, p. 1-17.
17. Llerena Jr JC, et al. Guidelines for the diagnosis, treatment and clinical monitoring of patients with juvenile and adult Pompe disease. Arq. Neuropsiquiatr. 2016; 74(2):166-176.
18. Tarnopolsky M, et al. Pompe Disease: Diagnosis and Management. Evidence-Based Guidelines from a Canadian Expert Panel. The Canadian Journal of Neurological Sciences. 2016; 43:472-485.
19. Smith BK, et al. Diaphragm Pacing as a Rehabilitative Tool for Patients with Pompe Disease Who Are Ventilator-Dependent: Case Series. Physical Therapy. 2016; 5:696-703.
20. Dubrovsky A, et al. Consenso argentino sobre enfermedad de Pompe de inicio tardío. Medicina. 2018; 78.
21. Werneck LC, et al . Muscle biopsy in Pompe disease. Arq. Neuro-Psiquiatr. 2013; 71(5):284-289.
22. Kishnani PS, Howell R. Pompe disease in infants and children. The Journal of pediatrics. 2004; 144(5):S35-S43.
23. Mullerfelber W, et al. Late onset Pompe disease: Clinical and neurophysiologicalspectrum of 38 patients including long-term follow-up in 18 patients. Neuromuscul Disord. 2007; 17:698-706.
24. Kishnani PS, et al. Early treatment with a glucosidade phaprolongs long-term survival of infants with Pompe disease. Pediatr Res. 2009; 66:329-335.
25. Chien YH, et al. Early detection of Pompe disease by newborn screening is feasible: Results from the Taiwanscreening program. Pediatrics. 2008; 122:e39-e45.
26. Oda E, et al. Newborn screening for Pompe disease in Japan. MolGenet Metab. 2011; 104:560-565.
27. Byerne BJ, et al. Pompe disease: design, methodology, and early findings from the Pompe Registry. Molecular genetics and metabolism. 2011; 103(1):1-11.
28. Mullender N, et al. A Dutch guideline for the treatment of scoliosis in neuromuscular disorders, Scoliosis. 2008; 3(14).
29. Van Der Beek NA, et al. Rate of disease progression during long-term follow-up of patients with late-onset Pompe disease. Neuromuscul. Disord. 2009; 19:113-117.
30. Saich R, et al. Is Newborn Screening the Ultimate Strategy to Reduce Diagnostic Delays in Pompe Disease? The Parent and Patient Perspective. International Journal of Neonatal Screening. 2020; 6(1): 1.
31. Paascual-Pascual S, et al. Guía clínica de la enfermedad de Pompe infantil. Rev Neurol. 2016; 63(6):269-79.
32. Van Der Ploeg AT, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10‐year experience. European journal of neurology. 2017; 24(6):768-e31.
33. Van Capelle CI, et al. Childhood Pompe disease: clinical spectrum and genotype in 31 patients. Orphanet journal of rare diseases. 2016; 11(1): 65.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()