Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 16(2) - Junho 2016
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
RELATOS DE CASO
Glicogenose tipo IV: Doença de Andersen
Glycogen storage disease type IV: Anderse's Disease
Anna Letícia de Cerqueira Campos Villardi1; Beatriz Araújo da Costa Soffe2; Clara Figueiredo Leal de Abreu1; Joanna Andrade da Costa1; Julia Tostes Calvo1; Marcos André Giffoni da Silva3; Ramona Alessandra Souza da Silva2
1. Graduação em Medicina Residente de Pediatria do Hospital Municipal da Piedade
2. Graduação em Medicina Médica Pediatra do Hospital Municipal da Piedade
3. Graduação em Medicina Médico Pediatra e Chefe da Pediatria do Hospital Municipal da Piedade
Endereço para correspondência:
Avenida Vice Presidente José Alencar, 1.515, bl. 2, apto. 908 Jacarepaguá
22775-033 - Rio de Janeiro - RJ
Resumo
OBJETIVO: Colocar em evidência a glicogenose do tipo IV, doença autossômica recessiva, caracterizada como erro inato do metabolismo. Atentar sempre ao possível diagnóstico, pois o quadro clínico é variável, apresentando sintomas inespecíficos. Importante frisar que uma terapêutica eficaz e rápida, junto ao acompanhamento conjunto com a Nutrição, a Endocrinologia e a Genética são fundamentais.
DESCRIÇÃO DO CASO: SDNL, lactente, 10 meses, sexo feminino, com quadro clínico de vômitos pós-prandiais, precedidos de náusea e associados à diarreia de coloração esverdeada. Não apresentou febre. Foi diagnosticada com desidratação grave, iniciada terapêutica e hidratação venosa, sendo solicitada internação hospitalar. Durante a internação, observaram-se hepatomegalia, criança irritadiça e parâmetros de crescimento e desenvolvimento muito inferiores aos adequados. Idade óssea era compatível a 3-6 meses. Foi realizada investigação para erro inato do metabolismo, fechando o diagnóstico para Doença de Andersen.
DISCUSSÃO: Diagnóstico é essencial, pois há grande risco de recorrência familiar (aproximadamente 25% para cada novo filho do casal), sendo fundamental o aconselhamento genético. Quanto mais precoce for o diagnóstico, menores são as chances de desenvolver sequelas graves, tanto físicas quanto cognitivas. Existem critérios e sinais que levam a pensar em Doença Metabólica Hereditária. A lactente em questão apresentava adequados parâmetros de crescimento até o 5º mês, quando estava em aleitamento materno exclusivo; porém, a partir do 6º mês, com complementação alimentar, apresentou declínio importante e considerável dos parâmetros, sendo classificada como muito baixo peso para a idade. O exame físico abdominal era doloroso, com hepatomegalia, extremamente irritadiça, notável atrofia muscular e atraso do desenvolvimento neuropsicomotor.
Palavras-chave: glicogenose, Doença de Andersen, crescimento, desenvolvimento, desnutrição.
Abstract
OBJECTIVE: We aim to highlight the type IV glycogen storage disease, autosomal recessive disease characterized as inborn errors of metabolism. We must always pay attention to possible diagnosis, because clinical presentation is variable, with nonspecific symptoms. Importantly, an effective and quick treatment, with the accompanying conjunction with nutrition, endocrinology and genetics are key.
CASE DESCRIPTION: SDNL, infant, 10 months, female, with clinical picture of post prandial vomiting preceded by nausea and diarrhea associated with greenish tinge. No known fever. Diagnosed with severe dehydration, started therapy and intravenous hydration, being required hospitalization. During hospitalization were observed hepatomegaly, irritable child and growth and developmental parameters much lower than appropriate. 3 to 6 months compatible bone age. Conducted research for inborn errors of metabolism, closing the diagnosis for Andersen's disease.
DISCUSSION: Diagnosis is essential because there is great risk of familial recurrence (approximately 25% for each new couple's son), and fundamental genetic counseling. The earlier the diagnosis, the lower the chances of developing serious consequences, both physical and cognitive. There are criteria and signs leading to think of Hereditary Metabolic Disease. The infant in question had appropriate parameters growth until the fifth month, when it was in exclusive breastfeeding, but from the sixth month, with food supplementation, presented important and significant decline of the parameters, with scores very low weight for age. Abdominal physical examination was painful, with hepatomegaly, extremely irritable, remarkable muscle atrophy and delayed psychomotor development.
Keywords: glycogen storage disease, glycogen storage disease type iv, growth, development, protein malnutrition.
INTRODUÇÃO
Os erros inatos do metabolismo são distúrbios genéticos e representam cerca de 10% dessas doenças, provocando falhas de síntese, degradação, armazenamento ou transporte de moléculas, que se manifestam em qualquer idade.1
A glicogenose do tipo IV (GSD IV) é uma doença autossômica recessiva; logo, irmãos afetados devem manifestar o mesmo subtipo de GSD IV, embora a idade de início e a clínica possam ser diferentes, devido à grande diversidade de sintomas.
Por ser decorrente de alterações de produção e consumo energéticos, provenientes de distúrbios do fígado, miocárdio, músculo e cérebro, pode cursar com hepatoesplenomegalia, miopatia (hipotonia e atrofia muscular) e retardo do crescimento, devido à ausência de glicogênio.3
Pode ser classificada em dois subtipos: (i) neuromuscular fatal perinatal - início no útero, diminuição dos movimentos fetais, polidrâmnio e hidropisia fetal, levando a óbito no período neonatal; e (ii) neuromuscular congênito começa no período de recém-nascido, com profunda hipotonia, desconforto respiratório e cardiomiopatia dilatada, com óbito na primeira infância.4
O objetivo deste relato de caso é mostrar a importância do diagnóstico precoce e do acompanhamento multidisciplinar, a fim de evitar sequelas futuras. Deve-se atentar aos sintomas básicos que podem ser os iniciais da doença.
DESCRIÇÃO DO CASO
SDNL, lactente, 10 meses, feminina, levada à UPA devido a quadro clínico de vômitos pós-prandiais, precedidos de náusea e associados a diarreia de coloração esverdeada. Não apresentou febre ou alteração urinária. Foi diagnosticada com desidratação grave, sendo iniciada terapêutica com Ceftriaxona e hidratação venosa (HV), sendo solicitada internação hospitalar.
Na internação em 12 de setembro de 2014, foi avaliada e obteve diagnóstico de gastroenterite aguda (GEA), desidratação grau III e atraso no desenvolvimento. Assim, foi suspensa a Ceftriaxona, mantida a HV e iniciada dieta com aleitamento materno (AM) complementado com papas doces e salgadas. Foram solicitados raios x (RX) de tórax, ultrassonografia (USG) de abdome e vias urinárias, exames laboratoriais, hemocultura, sorologias para toxoplasmose, rubéola, citomegalovírus, herpes vírus, anti-HIV, VDRL, pesquisa rotavírus, exame parasitológico de fezes (EPF), urinocultura e EAS, que, posteriormente, mostraram resultados negativos, com exceção do anti-HBS positivo e citomegalovírus IgG positivo. História imunológica deficiente de VIP e pneumocócica.
Na história alimentar, relato de AM exclusivo até o 6º mês e AM complementado com papas doces e salgadas; porém, não aceitava bem a papa doce, e os alimentos precisavam ser batidos, devido à dificuldade de deglutir e episódios de engasgo com pedaços de comida. Na história patológica pregressa, o perímetro cefálico (PC) era normal até o 5º mês; comprimento baixo para idade a partir do 5º mês; peso baixo para idade a partir 4º mês e muito baixo a partir do 6º mês.
Ao exame físico, contataram-se: peso de 5.045g; estatura de 61cm; PC de 43cm; frequências respiratória de 40irpm e cardíaca de 120bpm; temperatura axilar de 36,2ºC. AR: MVUA sem ruídos adventícios; ACV: RCR 2T, BNF, sem sopros. Abdome: peristáltico, fígado palpável a 7,5cm do rebordo costal direito (RCD) e 6cm apêndice xifoide, presença de hérnia umbilical.
No 4º dia de internação, mantinha quadro de diarreia, com média de dois episódios/dia, não apresentando mais vômitos e exame físico mantido. A USG abdominal evidenciou hepatomegalia de contornos regulares e textura homogênea, a vesícula biliar não foi visualizada. RX de ossos longos não mostrou alteração, inclusive compatíveis com hipovitaminoses; RX para avaliação da idade óssea foi compatível de 3 a 6 meses. Ecocardiograma feito no 6º dia de internação foi normal.
No 11º dia de internação, houve resolução da GEA; porém, mantinha-se irritada durante toda a internação, com abdome doloroso à palpação, além do fígado palpável a 6cm RCD, o espaço de Traube sempre foi livre. Havia atrofia muscular e atraso nos marcos do desenvolvimento, pois não sustentava a cabeça ou sentava. Solicitou-se parecer em serviço especializado em distúrbios do crescimento e desenvolvimento, devido à suspeita de erros inatos do metabolismo (EIM).
No 16º dia de internação, houve avaliação da nutrição com relato de baixa aceitação da dieta, mesmo após ajustes anteriores de consistência e preparo. Foi ofertado Infatrini, dieta enteral com alto valor proteico-calórico para lactentes, mas houve recusa. Devido ao quadro de acentuada desnutrição e perda ponderal considerada, sugeriu-se terapia nutricional via parenteral, objetivando suprir as necessidades proteico-calóricas.
No 18º dia de internação, apresentou dois picos febris (máximo de 38,2ºC). Permanecia chorosa e com hepatomegalia. Solicitaram-se RX de tórax, exames laboratoriais, coprocultura, com resultado negativo para pesquisa de Salmonella, Shigella e Escherichia coli enteropatogênica, EAS, EPF, hemocultura e rastreio para EIM. Nesse dia, iniciou-se alimentação via enteral, por sonda nasogástrica, administrando Infatrini concomitante à dieta via oral (almoço e jantar). Após 22 dias, suspendeu-se a dieta enteral. Ao exame abdominal, não havia dor à palpação superficial e profunda, mas o fígado permanecia a 5cm do RCD e a 4cm do apêndice xifoide; elspaço de Traube era livre.
A internação durou 41 dias, mas, no dia 24 de outubro de 2014, foi à consulta com especialista, na qual realizou novo rastreio para EIM, devido à forte suspeita diagnóstica. Decidiu-se pelo acompanhamento nesse serviço, concomitantemente à nutrição e à neurologia.
DISCUSSÃO
EIM são patologias genéticas de diversas manifestações clínicas, cujos sintomas iniciais ocorrem no período neonatal ou na infância, afetando um em cada 1.000 recém-nascidos vivos. Seu diagnóstico é essencial, pois há um grande risco de recorrência familiar de, aproximadamente, 25% para cada novo filho do casal, sendo fundamental o aconselhamento genético.4 Quanto mais precoce for o diagnóstico, menores são as chances de a criança desenvolver sequelas graves, tanto físicas quanto cognitivas. Existem critérios e sinais que, sem outra causa definida, levam a pensar em uma doença metabólica hereditária, sendo: hipotonia, hipoglicemia, irritabilidade, acidose, distúrbio hidroeletrolítico; crianças que, em associação, apresentem odores peculiares ou dismorfias; perda de habilidades adquiridas anteriormente; história de recorrência familiar ou consanguinidade entre os pais.1
As glicogenoses ocorrem devido a defeitos enzimáticos na via de degradação do glicogênio; a GSD IV tem maior repercussão no fígado, provocando hepatomegalia e hipoglicemia como sintomas principais. Nas primeiras horas de vida, os neonatos costumam ser assintomáticos; porém, há um período crítico evidenciado por volta de 6 a 8 meses, quando o lactente inicia alimentação complementar, pois passa a conter mais proteínas e a associar-se com longos períodos de jejum noturno.5
A paciente em questão abriu o quadro clínico com vômitos, náuseas e diarreia, associados a desidratação grave, desnutrição grau III e atraso do desenvolvimento. Apresentava adequados parâmetros de crescimento (peso, comprimento e PC) até o 5º mês, quando estava em AM exclusivo.
Todavia, a partir do 6º mês, quando houve complementação da alimentação com papas doce e salgada, apresentou declínio importante e considerável dos parâmetros, chegando a ser classificada como muito baixo peso para a idade (peso de 5.045g; comprimento de 61cm; PC de 43 cm aos 10 meses). Enquanto o esperado deveria ser em torno de 8kg para o peso; 70cm de comprimento e 46cm de PC (Gráfico 1)
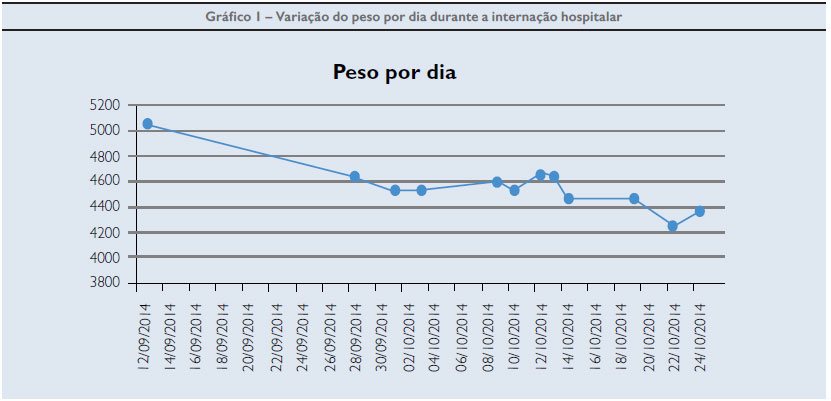
Na internação, o exame físico do abdome era doloroso, com hepatomegalia (7,5cm RCD e 6 cm do apêndice xifoide) e espaço de Traube livre. Era extremamente irritadiça, com notável atrofia muscular e atraso do desenvolvimento neuropsicomotor, visto que aos 10 meses não sustentava o pescoço e não sentava, havendo relato de frequentes de engasgos com a comida na forma sólida. O diagnóstico de doenças de armazenamento de glicogênio é um processo complexo. As formas hepáticas podem cursar com crise metabólica aguda, hipoglicemia, acidose lática e desidratação severa.2
As provas de função hepática e a dosagem de ácido úrico estão comumente elevadas. Nos exames laboratoriais, mostravam-se alterados TGP, TGO, fosfatase alcalina, gama GT, ácido úrico, bilirrubina total e bilirrubina direta, albumina e proteínas totais (Tabela 1).
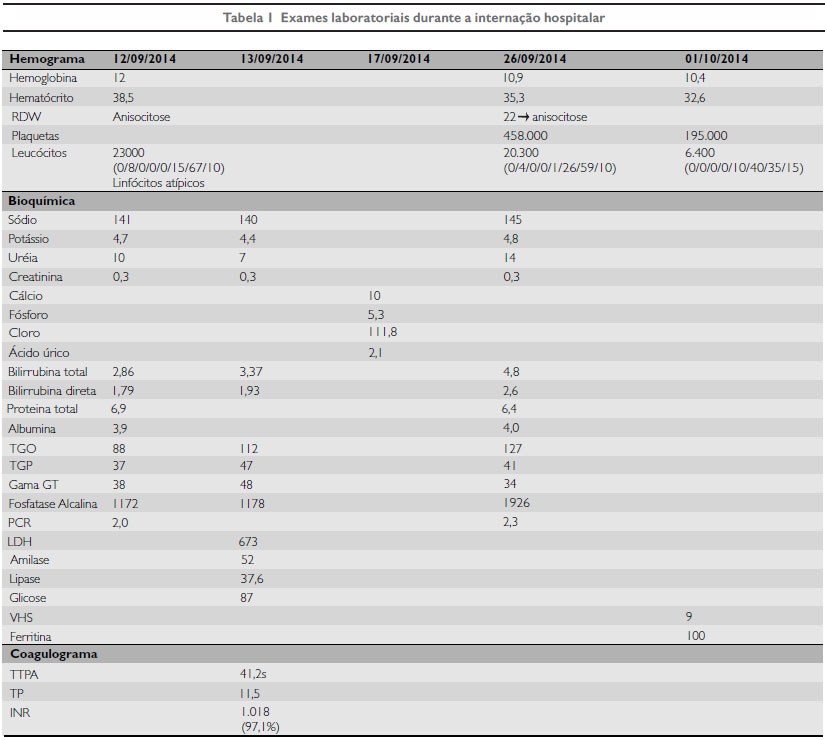
A glicose e os eletrólitos eram normais. No hemograma, havia anemia, anisocitose e leucocitose com linfócitos atípicos. A confirmação do diagnóstico requer escalonamento de testes, frequentemente invasivos, com jejum controlado em ambiente hospitalar com coleta regular de insulina, glicemia, lactato e eletrólitos, podendo durar até 16h. Como terapêutica, foram realizados HV e uso de nutrição enteral com Infatrini ao longo da internação, devido à não aceitação da dieta associada à grave desnutrição proteico-calórica. Os exames de ecocardiograma e USG de abdome estavam normais. Foi realizado RX de punho esquerdo para avaliação de idade óssea, a qual foi compatível com 3 a 6 meses. A paciente fez acompanhamento nos serviços de puericultura, de crescimento e desenvolvimento e nutrição.
Alguns sinais e sintomas são sugestivos de EIM, portanto, deve-se sempre ter atenção aos quadros clínicos que cursem com letargia ou coma, vômitos, retardo do crescimento e desenvolvimento, hipotonia, recusa alimentar e hepatoesplenomegalia. Devem ser sempre lembrados como diagnósticos diferenciais, visto que uma terapêutica eficaz e em intervalo de tempo adequado pode fazer muita diferença na evolução desses casos. É fundamental o acompanhamento conjunto com a Nutrição, a Endocrinologia e a Genética.
REFERÊNCIAS BIBLIOGRÁFICAS
1. El Husny AS, Cadalto MCF. Erros inatos do metabolismo: revisão de literatura. Revista Paraense de Medicina. .2006; 20(2): 42-45.
2. Jardim LB, Ashton-Prolla P. Erros inatos do metabolismo em crianças e recém-nascidos agudamente enfermos: guia para o seu diagnóstico e manejo Jornal de Pediatria 1996.
3. Portal Educação. Glicogenoses: doenças de armazenamento de glicogênio - Artigo por Colunista Portal - Educação - terça-feira, 1 de janeiro de 2008 Avaliable from: http://www.portaleducacao.com.br/enfermagem/artigos/705/glicogenosesdoencas-de-armazenamento#ixzz3pz4Yr57S
4. Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, et al. Glycogen storage disease Type IV. Seattle (WA): University of Washington 2013.
5. Ferreira JP et al. Pediatria: diagnóstico e tratamento; SPRS, capítulo 81, p. 768-78.
6. Nelson. Tratado de pediatria. 18ª edição, parte X, capítulo 84, p. 527-28.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()