Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 15(2) - Setembro 2015
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
- Outros do Autor
RELATOS DE CASO
Síndrome de Wiskott-Aldrich: Relato de caso
Wiskot-Aldrich Syndrome: Case report
Pedro Jose Secchin de Andrade
Mestre em Medicina Tropical pelo Instituto Oswaldo Cruz - Fiocruz (Médico)
Endereço para correspondência:
Hospital Municipal Rocha Maia
Praia de Botafogo, 460, apto 841
Botafogo - Rio de Janeiro - RJ. CEP: 22250-040
Resumo
OBJETIVO: Reconhecimento da Síndrome de Wiskott-Aldrich (SWA) pelos pediatras para abordagem e tratamento adequado da doença.
DESCRIÇÃO DO CASO: Criança de 6 anos, masculino, com SWA, atendido na emergência com história de mordida e arranhadura de cão doméstico. Paralelamente, também apresentava quadro de eczema em dorso do pé direito.Foi feito transfusão de concentrado de plaquetas, imunoglobulina humana, soro antirrábico, amoxicilina-clavulanato e observação do cão. O paciente evoluiu de forma estável.
DISCUSSÃO: A SWA é uma imunodeficiência primária causada por mutação no gene WASP localizada no cromossomo X.As manifestações clínicas da síndrome incluem:trombocitopenia com tamanho de plaquetas reduzido, infecções recorrentes, presença de eczema e aumento da incidência de manifestações autoimunes e neoplasias. A sobrevida desses pacientes é baixa, e o tratamento se constitui em antibioticoterapia e reposição de imunoglobulinas, mas somente o transplante de células hematopoiéticas é curativo.
Palavras-chave: Síndrome de Wiskott-Aldrich, Síndromes de Imunodeficiência, eczema.
Abstract
p>OBJECTIVE: Recognition of Wiskott-Aldrich Syndrome (WAS) by pediatricians for proper treatment of the disease.CASE DESCRIPTION: In this article, we report a case of a 6-year-old male with WAS and history of domestic dog bite and scratch. In addition, he also had eczema in the back of right foot. The treatment consisted of platelet transfusion, human immunoglobulin, anti-rabies serum and amoxicillin-clavulanate; the dog was also put under observation.The patient progressed with a good response.
DISCUSSION: The WAS is a primary immunodeficiency caused by mutations in the WASP gene located on X-chromosome. Clinical manifestations of the syndrome include: thrombocytopenia with small platelet size, recurrent infections, eczema and increased incidence of autoimmune manifestations and neoplasms. Patient survival is poor, and the treatment is based on antibiotic therapy and immunoglobulin replacement, but only hematopoietic cell transplantation is curative.
Keywords: Wiskott-Aldrich Syndrome, Immunologic Deficiency Syndromes, eczema
RELATO DO CASO
Paciente masculino, 6 anos, branco, portadora da Síndrome de Wiskott-Aldrich (SWA), foi à emergência apresentando lesão tumoral equimótica na região infraorbitária esquerda; equimose e escoriação em lábio inferior (Figura 1) e duas equimoses em região cervical anterior (Figura 2), decorrente de mordida e arranhadura há 2 dias por cão doméstico vacinado. Linfadenomegalia cervical pouco dolorosa à palpação. Bom estado geral e afebril. Braço esquerdo com duas manchas de 1,5 cm de diâmetro de cor violácea (Figura 3). Paralelamente, também apresentava quadro de eczema, com a presença de pápulas eritematosas pruriginosas, crostas, de início há 3 anos, em dorso do pé direito (Figura 4). Desenvolvimento físico-psíquico-motor sem anormalidades, porém, com infecções recorrentes. Hematócrito: 37,3%, leucócitos: 6.100mil/mm3, linfócitos: 12,3%, neutrófilos: 65,7%, plaquetas: 11.000mil/mm3. Os pais e um irmão são saudáveis, entretanto, o pai alega que teve outro filho que faleceu aos 6 anos por complicações da mesma síndrome. Foi feito transfusão de concentrado de plaquetas, imunoglobulina humana, soro antirrábico, amoxicilina-clavulanato e observação do cão. O paciente evoluiu com quadro estável, sem inter-corrências, e acompanhado em serviço especializado.
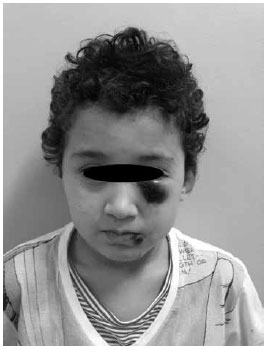
Figura 1 - Equimoses em região periorbitária esquerda e no lábio inferior.
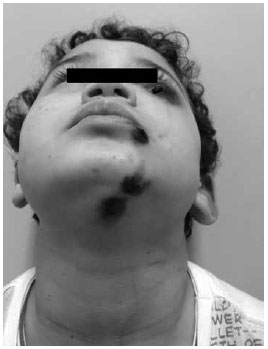
Figura 2 - Presença de equimoses em regiões do mento, lábio inferior e periobitária esquerda.
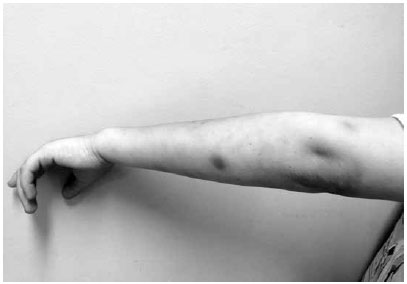
Figura 3 - Equimoses em braço esquerdo.

Figura 4 - Presença de lesão hiperemiada, escoriações, pápulas e crostas em dorso de pé direito.
DISCUSSÃO
O caso descrito é de uma criança portadora da Síndrome de Wiskott-Aldrich, uma imunodeficiência primária causada por mutação no gene WASP (proteína da síndrome de Wiskot-Aldrich), localizada no braço curto do cromossomo X (Xp11.22-11.2384)1,2. O fenótipo clínico é variável com o tipo da mutação2,3 e as formas de apresentação clínica são: a clássica, a trombocitopenia ligada ao X e a neutropenia congênita ligada ao X1. A Síndrome de Wiskott-Aldrich é rara, com a incidência de 1 a 10 em 1 milhão de indivíduos4.
O padrão clássico de manifestações clínicas da SWA inclui: trombocitopenia com tamanho de plaquetas reduzido, infecções recorrentes devido à deficiência da função dos linfócitos T e B, presença de eczema persistente e aumento da incidência de manifestações autoimunes (anemia hemolítca autoimune, vasculite, glomerulonefrite, púrpura de Henoch-Schöenlein e doença infamatória intestinal) e neoplasias (leucemia, linfoma e mielodisplasias)1,2,5,6. Os níveis de IgA e IgE estão elevados e os de IgM baixos.
O eczema se desenvolve em 80% dos pacientes, aparecendo, em geral, no primeiro ano de vida. Pode se apresentar desde a forma mais branda e localizada, até as mais graves. Predispõe a infecções cutâneas bacterianas e virais e a terapia requer hidratação da pele e corticoide tópico e, se indicado, sistêmico1,2,7.
A trombocitopenia e plaquetas de tamanho reduzido são os achados clínicos mais característicos para suspeita da síndrome. A confirmação do diagnóstico se dá pela diminuição ou ausência da proteína SWA nas células sanguíneas ou através da presença de uma mutação no gene WASP2. A história familiar também deve ser investigada2.
Antibioticoterapia adequada e reposição de gamaglobulina são medidas que aumentam a expectativa de vida dos doentes2,4,8. As transfusões de plaquetas podem ser usadas em algumas situações de hemorragias e o transplante de células-tronco hematopoiéticas, do sangue do cordão umbilical ou de medula óssea, é a única terapia curativa. Não são recomendadas vacinas com vírus vivos1,4.
Os pacientes, geralmente, sobrevivem até os 20 anos e as principais causas de morte são: infecções (44%), hemorragias (23%) e neoplasias malignas (26%)9.
A realização do diagnóstico precoce e diferencial e o manejo adequado podem evitar complicações graves nos doentes, reduzindo assim a morbimortalidade relacionada à doença e aumentando a qualidade de vida dos pacientes.
AGRADECIMENTOS:
Ao Felipe Dalvi Garcia pela correção do manuscrito.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Ochs HD, Filipovich AH, Veys P, Cowan MJ, Kapoor N. Wiskott-Aldrich Syndrome: Diagnosis, Clinical and Laboratory Manifestations, and Treatment. Biol Blood Marrow Transplant. 2009;15:84-90.
2. Immune Deficiency Foudation [Internet]. Wiskot- Aldrich Syndrome [Acesso 09 ago 2014]. Disponível em http://primaryimmune.org/about-primary-immunodeficiencies/specific-disease-types/wiskott-aldrich-syndrome/
3. Bostcardo M, Marangoni F, Aiut A, Villa A, Roncarolo MG. Recent advances in understanding the pathophysiology of Wiskot-Aldrich syndrome. Blood. 2009;113(25):6288-95.
4. Ochs HD, Thrasher AJ. The Wiskott-Aldrich syndrome. J Allergy Clin Immunol 2006;117:725-38.
5. Sullivan KE, Mullen CA, Blaese RM, Winkelstein JA. A multiinstitutional survey of the Wiskott-Aldrich syndrome. J Pediatr 1994;125:876-85.
6. Trasher AJ. New insights into the biology of Wiskot-Aldrich syndrome (WAS). Hematology. 2009;132-8.
7. Stehm ER, Ochs HD, Winkelstein IA, eds. Immunologic Disorders in Infants and Children. Fifh editon. Philadelphia: Saunders; 2004.
8. Consenso Brasileiro sobre o uso de imunoglobulina humana em pacientes com imunodeficiências primárias. Rev bras alerg imunopatol 2010;33:104-116.
9. Souza MS, Amaral SMM. Síndrome de Wiskott-Aldrich: relato de caso. Residência Pediátrica 2011;1(2):19-25.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()