Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 7(2) - Outubro 2006
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
- Outros do Autor
Revisoes em Pediatria
Linfangiectasia pulmonar na infância
Aline Liporrage do Amor Divino Nogarol
Ex-especilizanda do curso de Especializaçao em Pneumologia Infantil do IPPMG-UFRJ.
Endereço para correspondência:
Instituto de Puericultura e Pediatria Martagão
Gesteira da Universidade Federal do Rio de
Janeiro (IPPMG-UFRJ)
Av. Brig Trompwsky s.n.
Cidade Universitária Ilha do Fundão
Cep 21941-590, Rio de Janeiro, RJ
Resumo
OBJETIVO: Descrever aspectos clínicos, radiológicos, histopatológicos e terapêuticos sobre linfangectasia pulmonar na infância, entidade rara, de reconhecimento difícil.MÉTODO: revisão da literatura através do banco de dados PubMed.
CONCLUSÃO: a linfangestasia pode se apresentar sob as formas primária (ou congênita) e secundária.No primeiro caso,pode ser de aparecimento precoce ou tardio,sendo,esta forma, de diagnóstico mais difícil que a anterior.A suspeição clínica e a realização de biópsia pulmonar na maioria das vezes selam o diagnóstico. O tratamento é muito limitado.
Palavras-chave: linfangiectasia pulmonar, malformaçao; congênita; criança
Abstract
OBJECTIVE: To describe clinical,radiological and histopatological and therapeutic features of the pulmonary lymphangectasis (PL) in children,a rare and a difficult to recognize disease.METHODS: a non systematic review based on Pubmed databank.
CONCLUSION: PL may be primary or congenital and secondary forms.The primary PL may be a early or late onset one; the last is more difficult to diagnose than the late PL. Clinical suspicion and pulmonary biopsy confirm the diagnosis. The treatment is very limited.
Keywords: pulmonary lymphangectasis; congenital lung malformations; child
INTRODUÇÃO:
A linfangiectasia pulmonar (LP) é uma condição rara na qual se observa proliferação e dilatação dos vasos linfáticos (1). Há dois tipos: a linfangiectasia pulmonar congênita (LPC) ou primária, provavelmente por desenvolvimento anômalo de vasos linfáticos pulmonares, e a secundária, como conseqüência de uma obstrução da drenagem linfática ou de uma cardiopatia congênita com obstrução do retorno venoso.(2)
A classificação mais utilizada para LP foi a proposta por Noonan e col.(3) e divide a LP em 3 grupos: grupo 1, fazendo parte de uma linfangiectasia generalizada, com hemihipertrofia e linfangiectasia intestinal; grupo 2, associada à cardiopatias congênitas que dificultem o retorno venoso pulmonar; grupo 3, isolada, por defeito primário no desenvolvimento de linfáticos pulmonares.
A LP também tem sido descrita em associação à outras alterações clínicas como a Síndrome de Noonan, asplenia, ictiose ou fazendo parte de uma nova condição autossômica recessiva, caracterizada por linfangiectasia pulmonar congênita, quilotórax, edema facial e de membros inferiores(4).
CLÍNICA:
A LPC limitada aos pulmões pode ser de inicio precoce ou tardio. A apresentação precoce é a mais comum e se caracteriza por desconforto respiratório importante com dispnéia e cianose no período neonatal, geralmente com óbito nas primeiras horas de vida é comum a presença de quilotórax.(5,6) Na maioria dos casos não há intercorrência durante a gestação e o parto. Seu diagnóstico não é fácil pois a clínica é bem semelhante à outras causas de desconforto respiratório em recém nascidos como doença da membrana hialina, algumas cardiopatias congênitas, atelectasia fetal, Mikity - Wilson. A presença de quilotórax aumenta o grau de suspeição e pela sua alta mortalidade não há tratamento descrito.
Na apresentação tardia os sintomas surgem no período pós-neonatal e sua evolução vai depender da extensão da doença pulmonar. A forma localizada geralmente acomete um ou dois lobos pulmonares com melhor prognóstico que a difusa uma vez que o tratamento é a ressecção do lobo ou lobos afetados.(7) A forma difusa é mais rara e há acometimento bilateral dos pulmões. Na literatura existem poucos casos de LPC isolada difusa e de início tardio(7,8,9). Todos apresentaram uma evolução bastante semelhante, com aparecimento de tosse, derrame pericárdico, quilotórax e infiltrado reticulo nodular difuso, com prognóstico muito ruim. Contrariamente, Barker e col.(2), em trabalho restropectivo, sugerem que a LPC difusa de início pós natal não apresente um prognóstico tão ruim quanto o descrito na literatura.
DIAGNÓSTICO:
O padrão radiológico clássico na LP é infiltrado reticulo nodular difuso, o que não permite o diagnóstico diferencial entre processos infecciosos pulmonares e cardiopatias congênitas congestivas. No entanto, a presença de infiltrado reticulo nodular difuso mantido que não melhora com uso de antibiótico ou diurético, é um fator de extrema importância para se pensar em LP. Linhas B de Kerley indicativas de linfáticos dilatados também podem ser encontradas (FIGURAS 1 e 2). Já a presença de derrame pleural é infreqüente, porém alguns casos podem apresentar derrame pleural de característica quilosa.(7,10,11) O aparecimento de pneumotórax é raro(12).
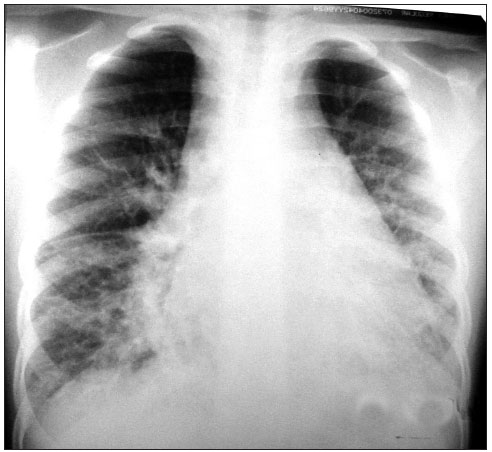
Figura 1 - Radiografia de tórax com infiltrado intersticial difuso bilateral e linhas B de Kerley.
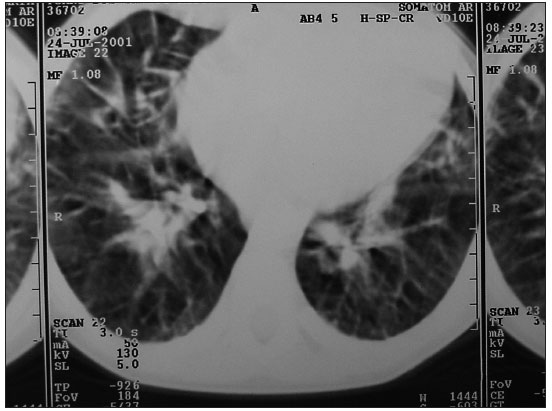
Figura 2: Tomografia computadorizada: espessamento do interstício pulmonar.
O diagnóstico de certeza é histopatológico através de biópsia pulmonar. O exame anátomo-patológico revela dilatação dos vasos linfáticos subpleurais, interlobares, perivasculares e peribronquicos 11 (FIGURA 3). Durante o período de investigação, muitas vezes o paciente é submetido à prova terapêutica para outras doenças pois o diagnóstico é pouco lembrado, pela sua raridade(11).
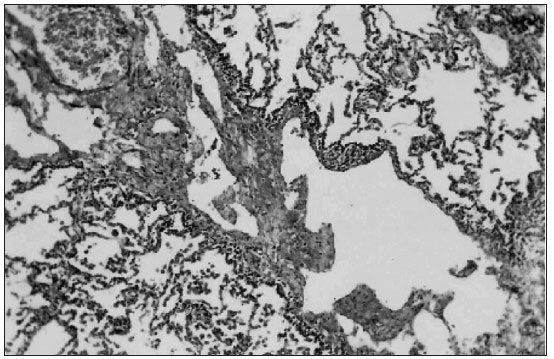
Figura 3. Exame histopatológico de biópsia pulmonar: dilatação dos vasos linfáticos subpleurais.
A linfografia deve ser realizada, não só para descartar obstrução linfática, como para auxiliar no diagnostico.
TRATAMENTO:
O tratamento da LPC isolada localizada é curativo com ressecção da área afetada. Já o da forma difusa é paliativo e tem como objetivo evitar o acúmulo de linfa nas serosas. Baseado nisto lança-se mão da terapia dietética com triglicerídeos de cadeia média ou nutrição parenteral pois a absorção se dá diretamente pelo sistema portal, o que leva à diminuição na formação de linfa. Diante da falência do tratamento conservador, lança-se mão do tratamento cirúrgico (pericardiectomia, pleurodese, circlagem da artéria pulomonar , ligadura do ducto torácico, shunt pleuroperitoneal, pleurectomia e abrasão pleural). Atualmente, os métodos mais utilizados em relação ao quilotórax são pleurectomia com pleurodese química associado ou não a ligadura de ducto torácico(11,13).
REFERÊNCIAS BIBLIOGRÁFICAS
1. Murray JF, Nadel JA , editors. Textbook of Respiratory Medicine. Philadelphia. W.B.Saunders; 2000:483.
2. Barker PM, Esther CR, Fordham LA, et al. Primary pulmonary lymphangiectasia in infancy and childhood. Eur Respir J 2004;24:413-9.
3. Noonan JA, Walters LR, Reeves JT. Congenital pulmonary lymphangiectasis. Am J Dis Child 1970;120:314-9.
4. Jacquemont S, Barbarot S, Bocèno M, et al. Familial congenital pulmonary lymphangiectasia, non-immune hydrops fetalis, facial and lower limb lymphedema: confirmation of Njostald's report. Am J Med Genet 2000;93:264-268
5. Gardner TW, Domm AC, Brock CE, et al. Congenital pulmonary lymphangiectasis. Clin Peditr(Phila). 1983;22(1):75-8
6. Brown MD, Reidbord HE. Congenital pulmonary lymphangiectasis. Amer J Dis Child 1967;114:654-7.
7. Diógenes MSB, Carvalho VB, Rozov T, et al. Pericardial effusion due to pulmonary lymphangietasis. Arq Bras Cardiol 1988;51(2):185-192.
8. Kelso JM, Kerr DJ, Lie JT, et al. Unusual diffuse pulmonary lymphatic proliferation in a young boy. Chest 1991;100(2):556-60.
9. Toltzis RJ, Rosenthal A, Fellows K, et al. Chylous reflux syndrome involving the pericardium and lung. Chest 1978;74(4):457-8.
10. Chung CJ, Fordham LA, Barker P, Cooper LL. Children with congenital pulmonary lymphangiectasis:after infancy. Am J Roentge 1999;173:1583-1588
11. Sant'Anna CC, Nogarol ALAD, March MF, Leal GM, Madi K, Alves L. A 4-year-old child with fever and persistent cough. Breathe 2006;2:269-272.
12. Siegal A, Katsenstein M,Wolach B. Neonatal pneumothorax, a rare complication of pulmonary cystic lymphangiectasis. Eur J Respir Dis 1985;66:153-157
13. Beghetti M, La Scala G, Belli D, et al. Etiology and management of pediatric chylotorax. J Pediatr. 2000;136(5):653-8
Trabalho realizado no Instituto de Puericultura e Peditria Martagão Gesteira da Universidade Federal do Rio de Janeiro (IPPMG-UFRJ).
AVALIAÇÃO
1. A respeito da linfangectasia pulmonar congênita cujo início se dá após o período neonatal, é correto dizer que cursa com:
a) linfangectasia intestinal e cardiopatia congênita
b) linfangectasia intestinal e edema facial
c) febre, taquicardia, derrame pleural e edema de membros inferiores
d) tosse, dispnéia, derrame pericárdico e quilotórax
2. Nos casos graves de quilotórax decorrente de linfangectasia pulmonar a melhor conduta é a ligadura do ducto torácico e:
a) dieta hipocalórica
b) pleurodese
c) circlagem da artéria pulmonar
d) citostáticos
Preencher ficha na página 28 e enviar à SOPERJ

![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()