Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 23(3) - Setembro 2023
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Relato de caso: a deficiência da adenilosuccinato liase
Case report: adenylosuccinate liase deficiency
Larissa Braz Gomes Vitoriano1; Lívia Gonçalves de Lima1; Luisa Longhini Martins1; Nathalia Destefano Ferreira1; Talita Russo Mini1; Tainá Regina Damaceno Silveira2; Charles Marques Lourenco1,2,3,4; Sheila Andrade de Paula Cecchetti1; Jaqueline Harouche Rodrigues Fonseca4
DOI:10.31365/issn.2595-1769.v23i3p96-101
1. Faculdade de Medicina - Centro Universitário Estácio de Ribeirão Preto - Ribeirão Preto - São Paulo - Brasil
2. Cetogene AG - Rostock - Meclemburgo-Pomerânia Ocidental - Alemanha
3. Faculdade de Medicina Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - São Paulo - Brasil
4. Laboratório - Diagnósticos laboratoriais especiais, - Rio de Janeiro - Rio de Janeiro - Brasil
Endereço para correspondência:
Recebido em: 14/07/2022
Aprovado em: 26/09/2023
Resumo
OBJETIVO: O artigo descreve um caso raro em criança brasileira diagnosticada com deficiência da adenilosuccinato liase (ADSL), destacando as características clínicas e evolução do quadro neurológico, a fim de estabelecer semelhanças diagnósticas que possam ser úteis aos pediatras e/ou neuropediatras no reconhecimento precoce dessa enfermidade. A mutação autossômica recessiva no gene ADSL, localizada no cromossomo 22, foi identificada por meio da técnica do sequenciamento genético. As informações apresentadas foram obtidas por meio de revisões de prontuário, laudos de exames de imagens, entrevistas com a mãe do paciente, registros audiovisuais e revisão de literatura.
RELATO DO CASO: Relata-se um caso da forma infantil clássica da deficiência da ADSL, com apresentação de epilepsia tardia, diagnosticado tardiamente após sequenciamento genético.
DISCUSSÃO E CONCLUSÃO: O caso relatado e publicações abordadas trazem uma reflexão quanto ao diagnóstico tardio em decorrência da falta de conhecimento sobre os principais sinais manifestados nos estágios iniciais de vida dos portadores da deficiência e a possibilidade de inclusão do gene ADSL entre os painéis de sequenciamento genético para todas as crianças que possuem atraso no desenvolvimento neuropsicomotor. Ademais, o conhecimento precoce da deficiência possibilitará um aconselhamento genético aos pais e melhor manejo terapêutico em relação à qualidade de vida das crianças enfermas, postergando, assim, a evolução das incapacidades através de tratamentos multidisciplinares.
Palavras-chave: Genética médica. Transtornos do Neurodesenvolvimento. Predisposição Genética para doença.
Abstract
OBJECTIVE: This article describes a rare case in Brazilian child diagnosed with adenylosuccinate lyase (ADSL) deficiency, highlighting the clinical characteristics and evolution of neurological conditions, in order to establish diagnostic similarities that may be useful to pediatricians and/ or neuropediatricians regarding the early recognition of this disease. Autosomal recessive mutation in the ADSL gene, located on chromosome 22, was identified using the gene sequencing technique. This research was performed through medical record analysis, imaging test reports, interviews with the patients' mother, audiovisual records and literature reviews.
CASE REPORT: We report a case of the classic infantile form of ADSL deficiency, with late-onset epilepsy, diagnosed late, after genetic sequencing.
DISCUSSION AND CONCLUSION: The case reported is associated with literature, bring a reflection on the late diagnosis due to the lack of knowledge of the main signs manifested in the early stages of life of people with disabilities and the possibility of including the ADSL gene in genetic sequencing panels for all children who have delays in their neuropsychomotor development. Furthermore, early knowledge of the disability will enable genetic counseling to parents and better therapeutic management in relation to the quality of life of sick children, to delay the evolution of disabilities through multidisciplinary treatments.
Keywords: Genetics. Neurodevelopmental Disorders. Genetic Predisposition to Disease.
Introdução
A adenilosuccinato liase (ADSL) é uma enzima envolvida em duas vias do metabolismo de nucleotídeos de purínicos.1 Em 1984, Jaeken e Van den Berghe2 descreveram, pela primeira vez, a deficiência de ADSL (OMIM #103050) em mais de 60 casos na Bélgica e Holanda.3,4 A deficiência de ADSL é causada por mutações no gene ADSL (OMIN #608222) autossômica recessiva, localizado no cromossomo 22.5 Já foram identificadas mais de 50 variantes no gene de ADSL, sendo a mutação do tipo missense R426H a mais comum.5
A incidência ainda é desconhecida na população mundial, por ser uma doença rara, existindo pouco mais de 90 indivíduos detectados com essa afecção.6
A enzima ADSL participa da via metabólica de biossíntese e da reciclagem dos nucleotídeos de purina em vários tecidos do corpo.7,8 Nesse sentido, a via metabólica biossintética dos nucleotídeos de purina consiste em 11 etapas com a conversão da ribose-5-fosfato em adenosina monofosfato (AMP) ou em guanosina monofosfato (GMP), como demonstrado na Figura 17. Desse modo, ressaltam-se duas etapas dessa via, não sequenciais, com a transformação do ribotídeo de succinilaminoimidazol carboxamida (SAICAR) em ribotídeo de aminoimidazol carboxamida (AICAr), e da transformação de adenilossuccinato (S-AMP) em adenosina monofosfato (AMP), ambas catalisadas pela enzima ADSL.7 Nos pacientes com deficiência de ADSL, os substratos SAICAR e S-AMP acumulam intracelularmente, sofrendo cada um, uma desfosforilação, transformando-se em ribosídeo de succinilaminoimidazol carboxamida (SAICAr) e em succiniladenosina (S-Ado), respectivamente, no qual depois dessa desfosforilação seguem para os fluidos extracelulares também ilustrado em Figura 1.9
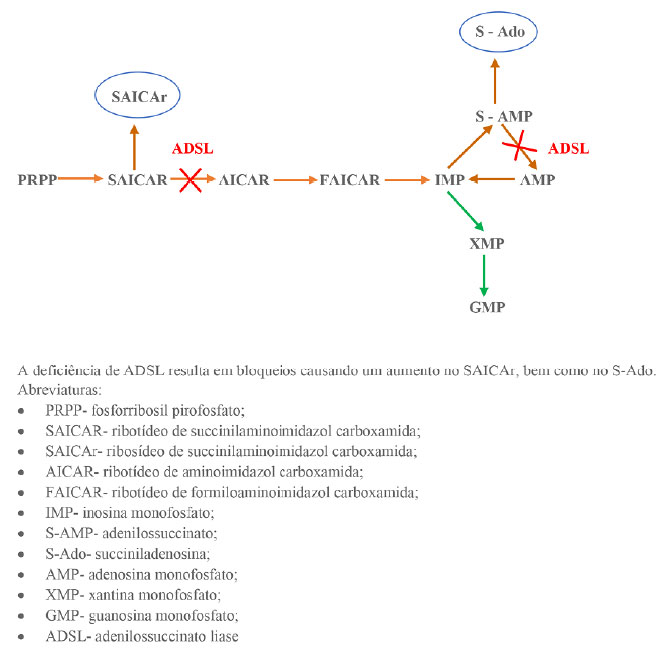
Figura 1. Esquema geral do metabolismo das purinas sendo influenciado pela adenilosuccinato liase. Fonte: Os autores (2021).
Clinicamente, é subdividida em quatro fenótipos clínicos:
• Fenótipo pré-natal - caracterizado por manifestações pré-natais, em que o embrião apresenta invariabilidade na frequência cardíaca, restrição no crescimento intrauterino, microcefalia e hipocinesia, levando a artrogripose e hipoplasia pulmonar.7,9
• Fenótipo neonatal fatal - a sintomatologia desde recém-nascido caracteriza por hipotonia neonatal (floppy baby), insuficiência respiratória, hipoplasia pulmonar, crises epiléticas intratáveis, evoluindo para óbito precoce.2,7,9
• Forma infantil clássica (ADSL do tipo I) - marcada por presença de retardo psicomotor grave, hipotonia axial e generalizada, parada do desenvolvimento e epilepsia neonatal aparecendo nos primeiros meses de vida.3,7,10
• Fenótipo atenuado (ADSL do tipo II) - forma leve a moderada surgindo mais tardiamente em relação à forma clássica, usualmente nos primeiros anos de vida; pacientes apresentam atraso neuropsicomotor, hipotonia, epilepsia e transtornos do espectro autista.2,7,10
Do ponto de vista diagnóstico, níveis anormalmente elevados dos substratos enzimáticos desfosforilados SAICAr e S-Ado são encontrados na urina, no líquido cefalorraquidiano (LCR) e no plasma sanguíneo. Esses metabólitos são indetectáveis nos biofluidos de pacientes saudáveis.9 Os metabólitos são detectados por técnicas de cromatografia líquida de alto desempenho (HPLC) e espectrometria de massas (HPLCMS). Para diagnóstico definitivo, porém, solicita-se o sequenciamento do gene ADSL.4 A deficiência de ADSL deverá ser suspeitada em caso de atraso inexplicado no desenvolvimento infantil, epilepsia de difícil controle e encefalopatia epilética de início neonatal.10
O artigo visa relatar o caso de um lactente brasileiro diagnosticado com deficiência de ADSL confirmado por identificação de mutação do gene ADSL por sequenciamento gênico, destacando características clínicas e evolução dos quadros neurológicos. Tal relato visa estabelecer características diagnósticas úteis para o reconhecimento precoce pelo pediatra ou neuropediatra, em vista do difícil acesso ao exame de urina e do líquido cefalorraquidiano, na rede pública e privada de saúde.
Relato de Caso
Lactente, sexo masculino, dezoito meses, filho de pais não consanguíneos, encaminhado para avaliação de encefalopatia epiléptica e atraso global de neurodesenvolvimento.
Durante o pré-natal, não houve intercorrências gestacionais. Nasceu de parto vaginal com bolsa rota por 30 minutos, pré-termo com 32 semanas, pesando 1,715kg (percentil 50), 50 cm de comprimento (acima do percentil 97), adequado para idade gestacional (AIG), perímetro cefálico 35 cm (acima do percentil 97) e APGAR 9 no primeiro minuto e 9 no quinto minuto.
Após o nascimento, houve a necessidade de hospitalização por 36 dias, sendo hospitalizado em unidade de terapia intensiva (UTI) neonatal, com desconforto respiratório e sepse precoce, permanecendo três dias em dispositivo de pressão positiva contínua nas vias aéreas (CPAP). Iniciou dieta enteral após 48 horas de vida, seguida por via sonda orogástrica e com um dia de vida, por via parenteral periférica. Posteriormente, recebeu antibioticoterapia durante sete dias desde o início do diagnóstico de sepse, além disso apresentou icterícia neonatal manejada com fototerapia de Bilitron por seis dias.
Obteve alta hospitalar realizando sucção débil e apresentando engasgos, sendo que a amamentação só foi efetivada em domicílio. Durante esse período, entretanto, os genitores observaram sinais de hipotonia.
Evoluiu com atraso nos marcos do desenvolvimento neuropsicomotor (DNPM), no qual, aos quatro meses de idade, paciente ainda apresentava hipotonia evidente, aos cinco meses desenvolveu sorriso social; aos nove meses de idade havia persistência da sucção débil; e com 12 meses de idade adquiriu a sustentação do segmento cefálico. Houve a iniciação com segmento neuropediátrico e fisioterapia motora, porém a mesma sem sucesso devido ao excesso de irritabilidade do paciente.
Com 19 meses, iniciaram-se as primeiras crises epilépticas, caracterizadas por movimentos oculares verticais aliados a movimentos mastigatórios e hipotonia corporal global, durando três minutos, de duas a três vezes no dia. A ressonância nuclear magnética (RNM) identificou sinais de redução volumétrica cerebral e eletroencefalograma (EEG) com descargas focais compatíveis com epilepsia focal.
Aos 27 meses, as crises epiléticas caracterizavam-se por serem tônico-clônicas em membros superiores e inferiores, durando cinco minutos com frequência de uma vez ao mês. Utilizou levetiracetam (3,87 mg/kg/dia) sem melhora, adicionada vigabatrina (193mg/kg/dia), obtendo melhora parcial, contudo frequentemente necessitava ser internado para manejo de crises epiléticas. Em seguida, realizou novo EEG, identificando encefalopatia difusa e epilepsia focal com múltiplos focos sem respostas paroxísticas na prova de fotoestimulação. Aos trinta meses, devido a episódio de piora súbita da hipotonia associada à parada de alimentação, é internado em UTI por suspeita de status epilepticus, posteriormente descartado. Manteve esquema de múltiplos anticonvulsivantes para controle de crises (Tabela 1).

Concomitante, o paciente apresentou irritabilidade excessiva junto a choro e agressividade, sem resposta a antipsicóticos (risperidona, cloridrato de clorpromazina e clonazepam). Ocasionalmente, evoluiu com extrema agitação, desperto por mais de 40 horas, com melhora do quadro clínico ao utilizar valproato de sódio e clonidina.
Após extensa investigação laboratorial e bioquímica, além da triagem para erros inatos do metabolismo como cromatografia de ácidos orgânicos urinários, perfil de acilcarnitinas em tandem, cromatografia quantitativa de aminoácidos plasmáticos, cariótipo com banda, cariótipo com banda G e SNP-array cromossômico, cujos resultados foram normais, optou-se pelo sequenciamento completo de exoma, demonstrando duas variantes missense em homozigose no gene ADSL: c.1277G>A (p. Arg426His), cada uma herdada de um dos genitores.
O exame morfológico revelou ausência de dismorfias craniofaciais significativas. Exame neurológico demonstrou motricidade ocular dentro da normalidade, contato visual limitado; força muscular aparentemente reduzida, hipotonia global, reflexos osteotendíneos vivos; sem alteração da sensibilidade; não emite sons específicos para comunicação ou sinais de lalação. Atualmente, não consegue se sentar sem apoio, engatinhar, andar ou falar e também não controla o esfíncter uretral ou retal.
Discussão
A deficiência de ADSL é uma doença progressiva autossômica recessiva rara que envolve o sistema nervoso central. Os sintomas variam de uma condição letal neonatal a progressões mais brandas, manifestada nos primeiros anos de vida. O amplo espectro clínico e a ausência de características patognomônicas levam a dificuldades no diagnóstico e diagnóstico diferencial da deficiência de ADSL.4
Os fenótipos epilépticos associados à deficiência de ADSL também são altamente variáveis, compreendendo mioclonia, crises parciais e generalizadas, espasmos infantis até o estado de mal epiléptico. A hipotonia generalizada no fenótipo mais grave da doença, costuma estar associada a hipertonia periférica. As crises epiléticas se iniciam no período neonatal ou após o primeiro o ano de vida. Metade dos pacientes com deficiência de ADSL apresentam epilepsia farmacorresistente, mas nem sempre associada à estado de mal epiléptico.11
O caso relato descreve um paciente com fenótipo precoce da doença, de encefalopatia epilética e do desenvolvimento. Ademais, exibe crises epiléticas tônico-clônicas generalizadas, crises epilépticas, hipotonia corporal global, presença de reflexos osteotendíneos, ausência de alterações na sensibilidade, atraso na linguagem e distúrbios oculares. O tratamento para as crises epiléticas do caso não obteve sucesso.
Características autistas em pacientes com deficiência de ADSL incluem falha no contato olho no olho, comportamento repetitivo, agitação, acessos de raiva e autoagressividade, além de movimentos das mãos, manipulação repetitiva de brinquedos, caretas, bater palmas, esfregar os pés, riso inadequado, balanço da cabeça e do tronco e sons estereotipados, sintomas que foram vistos no paciente em questão.12
No âmbito comportamental, há anormalidades como irritabilidade e agressividade e o atraso do DNPM, linguagem e ausência de autonomia, não havendo melhora com os tratamentos efetuados.13
Outras dismorfias como braquicefalia, occipital plano, suturas metópicas proeminentes, estrabismo divergente intermitente, nariz pequeno, filtro longo e liso, o lábio superior fino e orelhas inseridas baixas foram relatados por Holder-Espinasse et al.,14 enquanto outros autores mencionaram macrostomia, dentes bem espaçados e mandíbula proeminente. Todavia, não foram diagnosticados dismorfias craniofaciais significativas no paciente apresentado.
A variante associada à doença mais comum, do tipo missense com troca do aminoácido arginina por histidina na posição 426 da proteína que leva à instabilidade da enzima, foi identificada em 40% dos pacientes. Observou-se que essa substituição recorrente de nucleotídeos ocorre em homozigose em cinco casos (três dos quais apresentando um fenótipo grave) e heterozigose em dois pacientes. Quando a mesma mutação está associada a outra mutação da classe 5, o fenótipo é moderado/leve, enquanto quando associado a uma nova variante, o fenótipo é grave. As mutações p.R396H, troca da arginina por histidina na posição 396 da proteína e p.Y114H, troca do aminoácido tirosina por histidina na posição 114 da proteína descritas como causadoras de efeitos funcionais graves, foram inesperadamente associadas a um fenótipo mais brando. Semelhante a relatórios anteriores.15
Ao realizar o sequenciamento completo de exoma, revelou-se presença de duas mutações missense em homozigose no gene ADSL, cada uma herdada dos genitores não consanguíneos do caso. Apesar de a mutação detectada no probando não estar relacionada ao fenótipo grave, tem correlação com genótipo-fenótipo. Dessa maneira, devido ao difícil diagnóstico por meio das informações insuficientes na avaliação clínica do paciente e dificuldade de reconhecer o quadro clínico devido a multiplicidade de sintomas e similaridade com outras patologias, muitos casos são diagnosticados tardiamente, tornando irreversível a evolução neurodegenerativa progressiva da doença.
Mesmo não existindo terapêutica eficaz até o momento, é importante realizar o teste genético, a fim de diagnosticar precocemente a doença e possibilitar melhor suporte clínico e aconselhamento genético em uma gravidez subsequente.
A deficiência de ADSL possui amplo um espectro de fenótipos de progressão lenta a rápida, no qual a maioria dos pacientes passa por desafios diagnósticos com uma série de testes de um único gene, usados para definir o quadro. A utilidade clínica do sequenciamento do exoma é a detecção de variantes raras em pacientes com fenótipo suspeito de ser relacionado a uma doença genética com padrão de herança mendeliana.
Em pacientes com manifestações inespecíficas como deficiência intelectual sem outras características, o genoma pode ser indicado. Além disso, o gene ADSL deve ser incluído em painéis de genes para pacientes com características de deficiência intelectual isolada, atraso no desenvolvimento psicomotor e hipotonia de etiologia não determinada. A identificação de variantes pode identificar o modo de herança e, portanto, riscos para os filhos subsequentes.15
A heterogeneidade genética de distúrbios neurometabólicos mendelianos e o impacto clínico variável de mutações em genes individuais representam obstáculos ao uso de testes genéticos orientados por "fenótipo". Mesmo com a disponibilidade de testes de microarranjos cromossômicos e painéis genéticos de doenças específicas, a taxa de diagnóstico em distúrbios do neurodesenvolvimento pediátrico é baixa. A introdução do sequenciamento de nova geração na investigação melhorou consideravelmente o diagnóstico nos pacientes.
Referências
1. Jurecka A, Zikanova M, Tykil-Szymanska A, Krijt J, Bogdanska A, Gradowska W, et al. Clinical, biochemical and molecular findings in seven Polish patients with adenylosuccinate lyase deficiency. Molecular Genetics and Metabolism. 2008; 98 (4): 435-442.
2. Jaeken, J., Van den Berghe, G. An infantile autistic syndrome characterized by presence of succinylpurines in body fluids. The Lancet. 1984 Nov 10;2 (8411): 1058-61.
3. Clamadieu C, Cottin X, Rousselle C, Claris O. Adenylosuccinate lyase deficiency: An unusual cause of neonatal seizure. Archives de Pédiatrie. Feb. 2008; 15 (2): 135-138.
4. Macchiaiolo M, Barresi S, Cecconi F, Zanni G, Niceta M, Bellacchio E, et al. A mild form of adenylosuccinate lyase deficiency in absence of typical brain MRI features diagnosed by whole exome sequencing. Italian Journal of Pediatrics. Aug. 2017; 65 (2017): 1-7.
5. Stone RL, Aimi J, Barshop BA, et al. A mutation in adenylosuccinate lyase associated with mental retardation and autistic features. Nat Genet. 1992. Disponível em: https://pubmed.ncbi.nlm.nih.gov/1302001/
6. National Organization for Rare Disorders [base de dados online]. Itália: Universidade de Roma La Sapienza. 2010 [acesso em 26 de maio 2021]. Disponível em: https://rarediseases.org/rare-diseases/adenylosuccinate-lyase-deficiency/
7. Jurecka A, Zikanova M, Kmoch S, Tylki-Szymanska. Adenylosuccinate lyase deficiency. Journal of Inherit Metabolism Disorders. Aug. 2015; 38: 231-242.
8. Pérez-Dueñas B, Sempere A, Campistol J, Alonso-Colmenero I, Díez M, González V, et al. Novel features in the evolution of adenylosuccinate lyase deficiency. European Journal of Pediatric Neurology 2012 (16): 343-348.
9. Mouchegh K, Zikánová M, Hoffmann FG, Kretzschmar B, Kühn T, Mildenberger E, et al. Apresentação fetal letal e neonatal precoce de adenilosuccinato liase. Journal Pediatric 2007 150:57-61.
10. Jurkiewicz E, Mierzewska H, Kusmierska K. Adenylosuccinate lyase deficiency: The first identified polish patient. Brain & Development. Oct. 2007;29 (9): 600-602.
11. Jurecka A, Zikanova M, Kmoch S, Tylki-Szymańska A. Adenylosuccinate lyase deficiency. J Inherit Metab Dis. 2015; 38 (2): 231-42.
12. Gitiaux C, Ceballos-Picot I, Marie S, Valayannopoulos V, Rio M, Verrieres S, Benoist JF, Vincent MF, Desguerre I, Bahi-Buisson N. Misleading behavioural phenotype with adenylosuccinate lyase deficiency. Eur J Hum Genet. 2009; 17 (1): 133-6.
13. Mastrogiorgio, G., Macchiaiolo, M, Buonuomo P, Bellacchio E, Bordi M, Vecchio D, et al. Caracterização clínica e molecular de pacientes com deficiência de adenilosuccinato liase. Orphanet J Rare Dis. 2021; 16:112.
14. Holder-Espinasse M, Marie S, Bourrouillou G, Ceballos-Picot, Nassogne I, Faivre MC, et al. Towards a suggestive facial dysmorphism in adenylosuccinate lyase deficiency? Journal of medical genetics. 2002; 39: 210-440.
15. Macchiaiolo M, Buonuomo PS, Mastrogiorgio G, Bordi M, Testa B, Weber G, Bellacchio E, Tartaglia M, Cecconi F, Bartuli A. Very mild isolated intellectual disability caused by adenylosuccinate lyase deficiency: a new phenotype. Mol Genet Metab Rep. 2020; 23(6).
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()