Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 22(4) - Dezembro 2022
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Lipofuscinose ceroide neuronal: o que é necessário saber. relato de caso e revisão da literatura
Neuronal ceroid lipofuscinosis: what you need to know. case report and literature review
Jessyca Thays Melo de Andrade Ramos; Fernanda Veiga de-Góes; Marcela Rodriguez de-Freitas; Naiana de Fátima Silva; Alessandra Augusta Barroso Penna-e-Costa
DOI:10.31365/issn.2595-1769.v22i4p166-173
Instituto Fernandes Figueira, Neurologia Infantil - Rio de Janeiro - Rio de Janeiro - Brasil
Endereço para correspondência:
Recebido em: 05/04/2022
Aprovado em: 06/10/2022
Resumo
INTRODUÇÃO: Lipofuscinoses ceroides neuronais (LCNs) são um grupo de doenças neurodegenerativas causadas pelo acúmulo de lipofuscina nos lisossomos. Apresentam quadro clínico variado e inespecífico, tendo como manifestações principais: declínio do desenvolvimento cognitivo e motor, perda visual, epilepsia e morte precoce. As LCNs são consideradas a principal causa de encefalopatia progressiva da infância. Há, atualmente, 14 subtipos de LCN com 13 genes associados, pois questiona-se a existência da LCN9, não geneticamente definida. Dentre estes, variantes patogênicas no MFSD8 ocasionam a lipofuscinose ceroide neuronal do tipo 7 (LCN7).
OBJETIVO: Relatar a evolução de um paciente com o diagnóstico de LCN do tipo 7, revisar o tema e sugerir um organograma de investigação para não especialistas.
RELATO DE CASO: Paciente do sexo masculino, pais consanguíneos, encaminhado por quadro de regressão do desenvolvimento caracterizada por perda visual, tetraparesia espástica e ataxia de tronco. Investigação diagnóstica evidenciou pesquisa de inclusão de lipofuscina em linfócitos positiva, confirmando o diagnóstico de LCN. Posteriormente, pesquisa molecular demostrou homozigose do MFSD8, estabelecendo o diagnóstico de LCN7.
DISCUSSÃO: A combinação de regressão neurológica associada a perda visual e epilepsia é a apresentação clínica clássica das LCNs. Quadros de regressão do desenvolvimento neuropsicomotor devem levantar suspeita diagnóstica de LCN, visando o manejo terapêutico multidisciplinar precoce e aconselhamento genético. O diagnóstico precoce, particularmente da LCN tipo 2, é importante uma vez que há atualmente tratamento específico.
Palavras-chave: Lipofuscinoses Ceroides Neuronais. Epilepsia. Cegueira.
Abstract
INTRODUCTION: Neuronal ceroid lipofuscinoses (NCL) are a group of neurodegenerative diseases caused by the accumulation of lipofuscin in lysosomes. They present a varied and non-specific clinical picture, with the main manifestations: decline in cognitive and motor development, visual loss, epilepsy and early death. LCN are considered the main cause of childhood progressive encephalopathy. There are currently 14 subtypes of LCN with 13 genes, because the existence of LCN9, which is not genetically defined, is questioned. Among these pathogenic variants in MFSD8 cause Neuronal Ceroid Lipofuscinosis type 7 (LCN7).
OBJECTIVE: To report the evolution of a patient diagnosed with lipofuscinosis type 7, review the topic and suggest an investigation flowchart for non-experts.
CASE REPORT: Male patient, consanguineous parents, referred at the age of 4 years and 8 months to the Child Neurology service to evaluate developmental regression characterized by visual loss, spastic tetraparesis and trunk ataxia. Diagnostic investigation showed positive research for the inclusion of lipofuscin in lymphocytes, confirming the diagnosis of LCN. Subsequently, molecular research showed homozygosity of MFSD8, establishing the diagnosis of LCN7.
DISCUSSION: The combination of neurological regression associated with visual loss and epilepsy is the classic clinical presentation. Pediatricians and pediatric neurologists should suspect LCN in patients with developmental regression aiming at multidisciplinary therapeutic management and genetic counseling. LCN7, considered a rare type of LCN initially described in the Turkish population, however, has a global distribution. Early diagnosis, especially for type 2 NCL, is important, as there is currently a specific treatment.
Keywords: Neuronal Ceroid Lipofuscinoses. Epilepsy. Blindness.
INTRODUÇÃO
As lipofuscinoses ceroides neuronais (LCN) (OMIM# 256730), também conhecidas como doença de Batten, constituem um grupo de doenças neurodegenerativas caracterizadas pelo acúmulo lisossomal de pigmentos lipídicos autofluorescentes, com diferentes padrões ultraestruturais.1,2 Enquanto grupo, sua prevalência é de 1:12.500 nascidos vivos. As principais manifestações clínicas são atraso da linguagem, disfunção cognitiva, regressão do desenvolvimento, epilepsia mioclônica, distúrbio de comportamento, degeneração retiniana ocasionando perda visual e morte precoce.2,3
A idade de surgimento dos primeiros sintomas é variável e pode ser utilizada na classificação das quatro principais formas clínicas de LCN: infantil, infantil tardia, juvenil e adulta.4 Também são conhecidas pelos seus epônimos, entretanto não muito utilizados.5
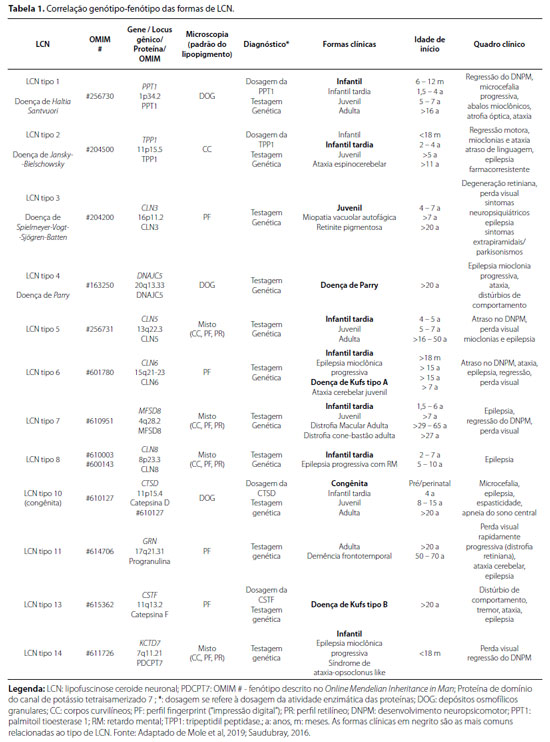
Os sintomas descritos nos fenótipos infantil (início entre 6 e 24 meses) e infantil tardio (início entre 2 e 5 anos) são a estagnação com posterior regressão do desenvolvimento neuropsicomotor (DNPM), epilepsia e perda visual. No fenótipo juvenil (início aos 5 a 7 anos), os sintomas iniciais geralmente são perda visual, mudanças de comportamento, regressão motora e epilepsia. No fenótipo adulto (início após os 16 anos de idade), as manifestações podem ser divididas em duas formas: epilepsia mioclônica progressiva geralmente sem perda visual (doença de Kufs tipo A) ou demência com regressão motora (doença de Kufs tipo B). Sinais e sintomas de ataxia, disfagia, mioclonias, coreia, tremores e distonia são especialmente evidentes nos fenótipos infantil e infantil tardio; enquanto sintomas de parkinsonismo são descritos na LCN tipo 3 juvenil.6 Atualmente, são conhecidos 13 genes relacionados aos subtipos de LCN2 (tabela 1), pois a LCN9 ainda não foi geneticamente definida. Destes genes, cinco codificam proteínas solúveis e os demais codificam proteínas transmembrana.5,6
O diagnóstico ocorre frequentemente em fases tardias da doença, pois os sinais e sintomas são inespecíficos.4 Os exames bioquímicos confirmam as LCNs tipo 1 e 2, com a dosagem enzimática da palmitoil tioesterase (PPT) e tripeptidil-peptidase 1 (TPP1), respectivamente. Os exames neurorradiológicos, como tomografia computadorizada (TC) e ressonância magnética (RM) do crânio, são importantes no diagnóstico diferencial.7 A análise de fibroblastos de pele ou linfócitos por microscopia eletrônica pode permitir a visualização de inclusões de pigmentos lipídicos autofluorescentes, confirmando assim o diagnóstico da LCN. Com a expansão e disponibilidade do sequenciamento de nova geração por meio de painéis genéticos ou exoma, o diagnóstico molecular específico do subtipo da LCN pôde ser realizado.8
O artigo objetiva revisar as LCNs, além de sugerir um organograma de investigação para crianças com transtornos neurodegenerativos, com o relato de um paciente com LCN do tipo 7 (OMIM# 611124) consequente à variante no MFSD8 (OMIM* 610951). Este trabalho foi submetido e aprovado pelo Comitê de Ética em Pesquisa de nosso hospital, com termo de consentimento livre e esclarecido assinado pelos responsáveis pelo paciente.
RELATO DE CASO
Paciente do sexo masculino, quarto filho de pais consanguíneos (primos de segundo grau) e irmãos saudáveis. Pré-natal foi adequando, com diagnóstico de doença hipertensiva gestacional. Nascido a termo, de parto cesáreo devido a descolamento de placenta, peso de 2.380 gramas, comprimento de 49 cm e 33 cm de perímetro cefálico, Apgar 8/9, recebendo alta no segundo dia de vida. Triagem neonatal, reflexo vermelho e emissões otoacústicas foram normais. O desenvolvimento motor decorreu normalmente (sentou sem apoio aos 6 meses, engatinhou aos 10 meses e andou aos 13 meses); entretanto, apresentou atraso de linguagem (primeiras palavras aos 15 meses, vocabulário com dez palavras aos 24 meses e emissão de frases somente após 4 anos).
O início dos sintomas se deu aos 4 anos e 8 meses, com regressão motora e de linguagem, e perda da marcha após dois meses. Foi avaliado no Serviço de Neurologia Infantil aos 4 anos e 10 meses, com exame neurológico evidenciando: dificuldade de compreensão e execução de ordens simples, atraso de linguagem, hipotonia oral, redução da acuidade visual, hipotonia de tronco com síndrome piramidal (tetraparesia espástica com fraqueza grau 4 -, hiperreflexia generalizada com clônus e sinal de Babinski), ataxia de tronco e movimentos anormais (mioclonias focais e generalizadas espontâneas). Durante a consulta, foram notados eventos de parada comportamental interpretados como crises epilépticas de início focal disperceptivas.
Os exames complementares inicialmente realizados foram: RM de crânio, evidenciando hipersinal em T2 e FLAIR nos tratos córtico-espinhais e substância branca profunda periventricular bilateral, discreto hipossinal em T2 em núcleos da base e tálamos e leve atrofia cortical difusa (Figuras 1a, 1b e 1c); eletroencefalograma (EEG) em sono com pontas bifocais nas regiões temporais médias; e potencial evocado visual normal.
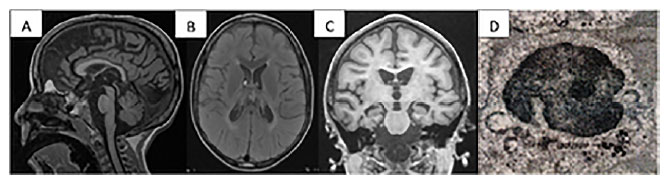
Figura 1. Exames Complementares do Paciente
Legenda: (A) sagital T1 3D evidenciando atrofia cerebelar com folias cerebelares e atrofia cortical; (B) T2 axial FLAIR com hiperintensidade de sinal periventricular e hipossinal em T2 em núcleos da base e tálamos; (C) coronal T1 com atrofia cortical difusa e hipocampo bilateralmente atrofiado; (D) pesquisa de inclusão de lipofuscina em linfócitos.
Foi iniciado tratamento medicamentoso com valproato de sódio (44 mg/kg/dia), pela suspeita de epilepsia focal e mioclonias não epilépticas.
Novos exames evidenciaram amônia e lactato normais; dosagem das enzimas: esfingomielinase, TPP1, PTT, hexoaminidases (A, A-MUGS), arilsulfatase A, quitotriosidade, ß-galactosidade e galactocerebrosidade, todas normais. A pesquisa de inclusão de lipofuscina em linfócitos (figura 1d) revelou inclusões constituída por depósitos eletrodensos, do tipo "fingerprint" e gotas lipídicas em linfócitos, confirmando o diagnóstico de lipofuscinose ceroide neuronal (Figura 1).
Com a disponibilidade da testagem molecular, foi realizado um painel genético revelando a presença de variantes em homozigose no gene MFSD8, confirmando o diagnóstico de LCN tipo 7.
A evolução clínica do paciente foi marcada por agravamento da epilepsia com crises mioclônicas, tônicas e clônicas diárias, necessitando de várias medicações anticrise [clobazam (1,5 mg/kg/dia); levetiracetam (70 mg/kg/dia); fenobarbital (5,5 mg/kg/dia); ácido valproico (45 mg/kg/dia); clonazepam (0,4 mg/kg/dia)]; além da associação de antipsicótico para o distúrbio do sono (clorpromazina). Os EEGs subsequentes demonstraram anormalidades irritativas multifocais. Com a deterioração do estado clínico, houve piora da tetraparesia espástica e do distúrbio de deglutição, evoluindo com desnutrição proteico-calórica, sendo necessária gastrostomia após episódio de broncoaspiração; veio a óbito aos 10 anos e 9 meses de idade.
DISCUSSÃO
As LCNs são um grupo de doenças hereditárias monogênicas, que se apresentam principalmente na primeira década de vida e correspondem às doenças neurodegenerativas mais comuns da infância. Caracterizam-se por epilepsia, perda visual e um progressivo declínio nas habilidades cognitivas e motoras.4
A classificação fenotípica das LCNs segundo a idade de apresentação clínica é dividida em: congênita, infantil, infantil tardia, juvenil e adulta.5 Outra classificação utilizada leva em consideração as variantes patogênicas em 13 genes diferentes (PPT1, TPP1, DNAJC5, CLN3, CLN5, CLN6, MFSD8, CLN8, CTSD, GRN, ATP13A2, CTSF e KCDT7), que são associadas a distintos subtipos e fenótipos clínicos. Em sua maioria possuem herança autossômica recessiva, exceto a LCN tipo 4, relacionada a um padrão de herança autossômica dominante6,8,9,10.
Em 1995, foi descoberto o gene associado à LCN tipo 1, PPT1, relacionado à inatividade da enzima lisossomal palmitoil proteína tioesterase (PPT1), que remove os resíduos de palmitato das proteínas.7 Na LCN tipo 2, o defeito subjacente é na enzima lisossomal tripeptidil peptidase (TPP1), um membro da família da serina carboxil proteinase. Os pacientes com LCN tipo 1 e 2 normalmente desenvolvem redução visual após o início dos sintomas neurológicos, enquanto em pacientes com LCN tipo 3 as manifestações visuais são os sintomas iniciais.1 A forma congênita é rara, exibindo microcefalia e epilepsia ao nascimento, resultando em morte nos primeiros dias de vida, ocasionada por variantes patogênicas em CTSD (LCN tipo 10). Na LCN forma adulta clássica, os sintomas geralmente começam por volta dos 30 anos, porém há relatos na adolescência ou idade adulta tardia. A doença de Kufs está relacionada a diversos genes (PPT1, CTSD, CLN3, CLN5, CLN6, CSTF, GRN) com herança autossômica recessiva, enquanto a doença de Parry (LCN tipo 4), muito rara, é correlacionada ao gene DNAJC5, de herança autossômica dominante. São descritas duas formas de doença de Kufs: tipo A (LCN tipo 6), manifestada por epilepsia mioclônica progressiva, enquanto o tipo B (LCN tipo 13) cursa com demência e sinais motores. O gene responsável pela LCN9 ainda não foi identificado, sendo questionada sua existência. O gene ATP13A2 é atribuído à LNC12, com início dos sintomas em torno de 8 anos: dificuldades de aprendizagem, instabilidade marcha, mioclonia, distúrbios do humor, e como acinesia, rigidez e fala disártrica. Tal gene também está relacionado à síndrome de Kufor-Rakeb, uma forma de parkinsonismo infanto-juvenil com demência, paralisia supranuclear e sinais piramidais.4,6
A LCN7 é causada por variantes patogênicas no gene MFSD8, localizado no cromossomo 4q28.1-q28.2, que codifica a proteína LCN7 ou MFSD8 (Major facilitator superfamily domain containing protein-8), localizada na membrana dos lisossomos com uma função transmembrana.11 Uma proteína não funcional impede o transporte adequado de moléculas pela membrana lisossomal e, consequentemente, leva ao acúmulo de macromoléculas e alteração do trânsito intracelular, desencadeando uma disfunção dos lisossomos e apoptose celular.11 O primeiro relato da LCN7 foi na Turquia, em 2004, em um estudo sobre o que se imaginava ser a LCN9, sendo a LCN7 inicialmente denominada variante turca.12 Estudos posteriores indicam incidência desta variante em outras populações13.
Em concordância com o caso relatado, as primeiras manifestações ocorrem entre 2 e 11 anos de idade (média aos 5 anos), sendo declínio motor e cognitivo, epilepsia, perda visual e ataxia os achados mais frequentes.12 Entretanto, é importante ressaltar que a perda visual nem sempre está presente na LCN7, como descrito em uma família egípcia, na qual a epilepsia se iniciou aos cinco anos de idade, seguida por rápido declínio cognitivo e motor, com morte precoce, porém sem regressão visual.13
Dos 25 pacientes de uma coorte turca com variantes patogênicas no MFSD8, 24 apresentaram o fenótipo infantil tardio, com média de idade apresentação aos três anos. Os sintomas mais frequentes foram: regressão do desenvolvimento (10/24; 42%) e epilepsia (9/24; 38%). A progressão da doença foi rápida, com regressão cognitiva, de linguagem e epilepsia. Perda visual ocorreu em 90% dos pacientes, enquanto ataxia e mioclonia, em 85%. A idade média de morte dos sete pacientes que faleceram foi 11,5 anos,12 porém o nosso paciente faleceu mais precocemente.
O diagnóstico das LCNs ainda é desafiador, uma vez que os sintomas iniciais podem ser confundidos com outras desordens neurodegenerativas. Os achados de ressonância magnética de crânio são inespecíficos, como vistos no paciente relatado, com hiperintensidade em T2 em substância branca profunda, periventricular ou porção posterior da cápsula interna, além da presença de atrofia cerebral, cerebelar e hipocampal.14
Na investigação inicial das LCNs, é sugerido o estudo enzimático, disponível apenas para as formas do tipo 1 e 2. Caso a dosagem enzimática seja normal, deve-se considerar outros tipos de LCNs, além de outras doenças neurodegenerativas.15
A pesquisa de inclusão de lipofuscina por microscopia eletrônica em fibroblastos, de pele ou de conjuntiva, ou linfócitos, foi por muito tempo considerada o exame padrão ouro para o diagnóstico. No entanto, são necessários profissionais experientes, nem sempre disponíveis, para realizar a diferenciação dos subtipos de LCN.16 A pesquisa de inclusão de lipofuscina em linfócitos foi realizada no paciente descrito. Com o menor custo e maior acessibilidade do sequenciamento de nova geração, vem sendo possível o diagnóstico mais precoce, além do esclarecimento do subtipo da LCN, por meio de painéis genéticos ou exoma.8
Para facilitar o raciocínio diagnóstico e exames a serem solicitados, propõe-se um fluxograma de investigação, de acordo com a disponibilidade dos exames complementares (Figuras 2a e 2b). No caso descrito, houve minuciosa investigação para doenças neurodegenerativas, inclusive para LCN2. A pesquisa de inclusões em linfócitos confirmou o diagnóstico de LCN, porém não foi determinado o subtipo, sendo este elucidado somente com o painel de epilepsia por sequenciamento de nova geração.
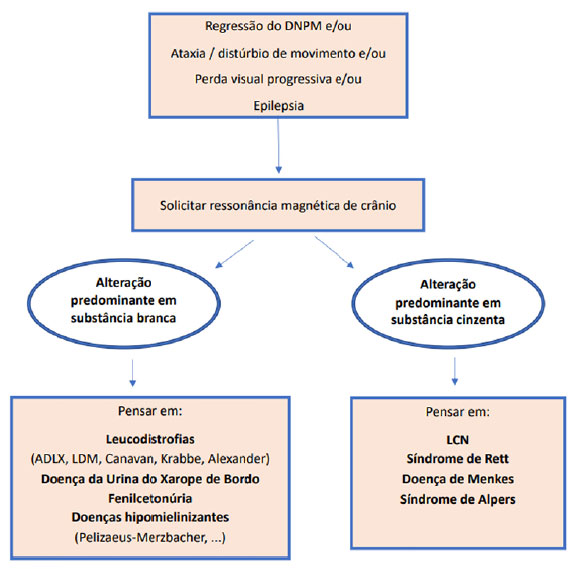
Figura 2a. Algoritmo diagnóstico para abordagem inicial de um paciente com regressão do desenvolvimento
Legenda: DNPM: desenvolvimento neuropsicomotor; ADLX: adrenoleucodistrofia ligada ao X.
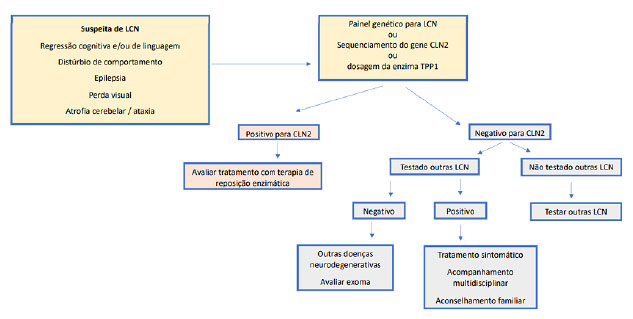
Figura 2b. Algoritmo diagnóstico para suspeita de LCN.
Ainda não há um tratamento preventivo ou curativo estabelecido para a maioria das LCNs. A reposição enzimática com a alfacerliponase (Brineura®) é uma opção terapêutica recente para o tratamento da LCN tipo 2.6,9 Há terapias gênicas em estudo (fase 1-2) para avaliar a segurança e eficácia de terapia gênica administrada via intratecal para LCN3, LCN6 e LCN7, entretanto ainda não disponíveis para utilização clínica.11,12 Há dados sugestivos do acúmulo lisossomal de glicoesfingolipídeo na LCN3 e LCN5, logo sendo aventada a possibilidade da prevenção de tal acúmulo com um inibidor da glicosilceramida sintase como o Miglustat®, já usado no Neimann-Pick tipo C. Recentemente, também foi demonstrado que a trealose (um dissacarídeo) promove a depuração lisossomal na doença de armazenamento por ativação de TFEB, um fator de transcrição envolvido na biogênese dos lisossomos e recrutamento. Há um estudo em aberto, com um pequeno número de pacientes LCN3, tentando combinar os benefícios potenciais de Miglustat e da Trealose usando um novo medicamento recentemente aprovado pelo FDA (Food and Drugs Administration) - BBDF101, que contém ambos os componentes.9,10 Houve apenas um estudo até agora, com o objetivo de esgotar o armazenamento endolisossomal na CLN1 pela combinação ação da fosfocistemaína (cliva a ligação tioéster numa proteína palmitoilada) e N-acetilcisteína (forte antioxidante que cliva a ligação tioster). O resultado no longo prazo mostrou pequenos benefícios subjetivos para os pacientes, juntamente com algumas melhorias relacionadas ao padrão de EEG e dissolução de armazenamento.9,10
Deste modo, no momento com a possibilidade concreta de tratamento para a LCN tipo 2 e tratamentos para outras formas de lipofuscinoses em estudo, torna-se importante a diferenciação do subtipo da LCN. Para a maioria das LCNs, o manejo é sintomático. As principais questões neurológicas a serem manejadas são:
- Epilepsia: em geral, é farmacorresistente, com múltiplos tipos de crise, predominando as mioclonias. As medicações anticrise mais eficazes são valproato de sódio e lamotrigina. Deve-se evitar o uso de carbamazepina, fenitoína e vigabatrina, pela possibilidade de piora das mioclonias. Como terapia adjuvante para as mioclonias refratárias, deve-se pensar em pregabalina, fenobarbital, benzodiazepínicos e, eventualmente, piracetam. O paciente relatado necessitou de cinco fármacos anticrise, com resposta parcial. O controle da dor também é um objetivo no tratamento, pois pode ser gatilho para as mioclonias.6
- Ataxia: é o sintoma clínico mais frequente da LCN2, LCN7, LCN6, LCN8 e da variante infantil tardia da LCN1. A ataxia é secundária ao envolvimento espinocerebelar. Atrofia cerebelar é um sinal neurorradiológico precoce, precedendo o alargamento dos sulcos corticais e dos ventrículos laterais.9,7
- Deterioração motora: fraqueza com perdas de marcos motores já adquiridos, acometimentos da musculatura ocular (estrabismo) e da deglutição (disfagia) por envolvimento do córtex motor e dos neurônios dos núcleos motores. Há o surgimento de espasticidade e alterações na marcha em diferentes formas de LCN, consideradas como um "marcador de progressão da doença".9,10 A espasticidade e imobilidade osteoarticular podem ser dolorosas e devem ser tratadas com fisioterapia motora, pregabalina, canabidiol e toxina botulínica local nos casos mais graves.6
- Declínio cognitivo: é característica marcante das LCNs. Na forma infantil tardia, a regressão no aprendizado escolar ocorre rapidamente, enquanto em pacientes "juvenis" a evolução é relativamente lenta.9 No entanto, na LCN2, muitas crianças apresentam um atraso na aquisição de habilidades cognitivas numa fase muito precoce, com atrasos mais evidentes na linguagem comparada com as habilidades motoras.9,10
- Distúrbios de comportamento: ocorrem no início do quadro da LCN2, LCN3 e LCN5, como um dos primeiros sintomas na idade escolar e em fases mais avançadas nas outras LCNs. Incluem: ansiedade, depressão, explosões de agressividade e manifestações psicóticas. Esses sintomas tendem a permanecer estáveis ou piorar ao longo dos primeiros anos, tornam-se menos significativos à medida que a doença avança e as habilidades funcionais e cognitivas são perdidas.9,10
- Perda visual: é um dos sintomas clássicos da LCNs, principalmente nos fenótipos de início na infância, ocorrendo de forma progressiva. Retinas, vias visuais e córtices visuais, neurônios ganglionares e células receptoras (cones e bastonetes) são envolvidos. A resposta retiniana prejudicada é visualizada pelo eletrorretinograma, e a tomografia computadorizada óptica permite o acompanhamento da degeneração progressiva da retina.
Apesar de a LCN ser uma doença rara e complexa, o pediatra geral possui papel primordial tanto na suspeita clínica de um paciente com regressão do desenvolvimento, como no acompanhamento conjunto com o especialista. A maioria das LCNs ainda não possui tratamento curativo, porém deve-se visar a melhor qualidade de vida e uso de sintomáticos dirigidos a queixas como: fraturas, trombose venosa e constipação e desnutrição.6 O manejo deve ser feito de maneira multidisciplinar com neuropediatra, pediatra geral e demais profissionais, de acordo com as necessidades do paciente.
O diagnóstico precoce pode possibilitar a terapia de reposição enzimática na LCN2 e nos demais tipos, sendo o melhor manejo terapêutico, com aconselhamento genético familiar.
REFERÊNCIAS
1. Lin J, Simão GN, Rodrigues MM. Erros Inatos do Metabolismo. In: Rodrigues MM, Vilanova LCP. Tratado de Neurologia Infantil.1 Ed. Rio de Janeiro: Atheneu, 2017. p. 568-746
2. Li W, Fan X, Zhang Y, Huang L, Jiang T, Qin Z, et al. A novel pathogenic frameshift variant unmasked by a large de novo deletion at 13q21.33-q31.1 in a Chinese patient with neuronal ceroid lipofuscinosis type 5. BMC Med Genet. 2020; 21(1):100.
3. Kohlschutter A, Schulz A, Bartsch U, Storch S. Current and Emerging Treatment Strategies for Neuronal Ceroid Lipofuscinoses. CNS Drugs. 2019; 33(4):315-325.
4. Mole S, Anderson G, Band H, Berkovic S, Cooper J, Holthaus SM, et al. Clinical challenges and future therapeutic approaches for neuronal ceroid lipofuscinosis. Pediatric Genetics & Genomics, Division of Biology and Biomedical Sciences. 2019; 18(1):107-116.
5. Vanier MT, Caillaud C, Levade T. Disorders of Spingolipid Synthesis, Sphingolipidoses, Niemman-Pick Disease Type C and Neuronal Ceroid Lipofuscinoses. In: Saudubray JM, Baumgartner RM, Walter J. Inborn Metabolic Diseases Diagnosis and Treatment. Berlim: 6a Ed. Springer; 2016 p.568-571
6. Mole SE, Schulz A, Haltia M. The neuronal ceroid-lipofuscinoses (Batten disease). In: Rosenberg RN, Pascual JM. Rosenberg's Molecular and Genetic Basis of Neurological and Psychiatric Disease. Londres: Ed. Elsevier; 2020 p.62-64
7. Gama R, Nakayama M, Távora D, Alvim T, Nogueira C, Portugal D. Lipofuscinose ceroide neuronal: achados clínicos e neurorradiológicos. Arq neuropsiquiatr. 2007;65(2A): 320-326.
8. Butz ES, Chandrachud U, Mole SE, Cotman SL. Moving towards a new era of genomics in the neuronal ceroid lipofuscinoses. Biochim Biophys Acta Mol Basis Dis. 2020;1866(9):165571.
9. Simonati A, Williams RE. Neuronal Ceroid Lipofuscinosis: The Multifaceted Approach to the Clinical Issues, an Overview. Front Neurol. 2022;13:811686.
10. Scarpa M , Bellettato CM, Sechi A. The Neuronal Ceroide Lipofuscinoses. In: Blau N, Vici CD, Ferreira CR, Vianey-Saban; van Karnebeek CDM. Physician's Guide to the Diagnosis, Treatment and Follow-Up of Inherited Metabolic Diseases. 2a Ed. Springer; 2022 p.1207-1234.
11. Kose M, Kose E, Ünalp A, Yılmaz Ü, Edizer S, Tekin HG, et al. Neuronal ceroid lipofuscinosis: genetic and phenotypic spectrum of 14 patients from Turkey. Neurol Sci. 2021;42(3):1103-1111.
12. Topçu M, Tan H, Yalnizoğlu D, Usubütün A, Saatçi I, Aynaci M, et al. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological and histopathologic studies. Turk J Pediatr. 2004;46(1):1-10.
13. Kousi M, Siintola E, Dvorakova L, Vlaskova H, Turnbull J, Topcu M, et al. Mutations in CLN7/MFSD8 are a common cause of variant late-infantile neuronal ceroid lipofuscinosis. Brain. 2009;132:810-819.
14. Biswas A, Krishnan P, Amirabadi A, Blaser S, Mercimek-Andrews S, Shroff M. Expanding the Neuroimaging Phenotype of Neuronal Ceroid Lipofuscinoses. AJNR Am J Neuroradiol. 2020;41(10):1930-1936.
15. Nita DA, Mole SE, Minassian BA. Neuronal ceroid lipofuscinoses. Epileptic Disord. 2016;18(S2):73-88.
16. Pérez-Poyato MS, Milà-Recasens M, Ferrer-Abizanda I, Cusí-Sánchez V, Vázquez-López M, Camino-León R, et al. Lipofuscinosis neuronal ceroidea: algoritmo diagnóstico y descripción clínica de las variantes infantil tardía finlandesa (CLN5) y turca (CLN7)]. Rev Neurol. 2012;54(9):544-50.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()