Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 22(4) - Dezembro 2022
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Raquitismo hipofosfatêmico ligado ao X: importância no diagnóstico diferencial no paciente pediátrico com déficit pôndero-estatural de causa desconhecida
X-linked hypophosphatemic rickets: importance in the differential diagnosis in pediatric patient with weight-height deficit of unknown cause
Larissa da Silva Chini1; Larissa de Menezes Cabral1; Marilene Ferraz Cavalieri1; Álvaro Martins Gabriel Isaac1; Zumira Aparecida Carneiro1; Laura Vagnini2; Thiago Hirose1; Charles Marques Lourenco1,2,3; Tainá Regina Damaceno Silveira4
DOI:10.31365/issn.2595-1769.v22i4p154-162
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - RIBEIRÃO PRETO - SP - Brasil
2. CPDP - Centro Paulista de Diagnóstico, Pesquisa e Treinamento - Medicina Diagnóstica, Genética Clínica - RIBEIRAO PRETO - SP - Brasil
3. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
4. Centogene AG, Genética Molecular - Rostock - Mecklemburgo-Pomerânia Ocidental. - Alemanha
Endereço para correspondência:
Recebido em: 03/06/2021
Aprovado em: 06/11/2022
Resumo
INTRODUÇÃO: O raquitismo hipofosfatêmico ligado ao cromossomo X é uma rara doença osteometabólica causada por mutações no gene PHEX (Endopeptidase Neutra Reguladora de Fosfato), localizado no cromossomo Xp22.11. Mutações nesse gene levam ao aumento dos níveis do fator de crescimento do fibroblasto 23 (FGF-23), prejudicando a absorção de fosfato nas células tubulares renais e no epitélio intestinal.
RELATO DE CASOS: Caso 1 Paciente feminino, 11 anos, encaminhada para investigação de déficit pôndero-estatural associado à deformidade óssea do membro inferior em varo; exames radiográficos foram compatíveis com alterações metafisárias sugestivas de raquitismo.
CASO 2 Paciente masculino, 8 anos, irmão do caso 1, encaminhado para avaliar quedas frequentes e dor em joelho ao deambular, apresentou achados radiográficos compatíveis com dolicocefalia, arqueamento dos membros inferiores em varo e displasia metafisária. Investigação bioquímica de ambos foi compatível com raquitismo hipofosfatêmico, posteriormente confirmado geneticamente com identificação de mutação patogênica no gene PHEX.
DISCUSSÃO: O raquitismo hipofosfatêmico ligado ao X constitui uma das principais formas de raquitismo de herança genética. Pacientes afetados usualmente manifestam sintomas ainda na infância, como atraso de crescimento estato-ponderal, mineralização óssea anormal, alterações dentárias, craniossinostose, além de osteomalácia e deformidades ósseas em membros inferiores. Diante dos avanços terapêuticos nessa área, é fundamental que o pediatra reconheça os sinais precoces dessa enfermidade, para que não só seja possível o correto e acurado manejo clínico, mas também a realização do aconselhamento genético familiar.
Palavras-chave: Raquitismo. Raquitismo Hipofosfatêmico. Raquitismo Hipofosfatêmico Familiar. Endopeptidase Neutra Reguladora de Fosfato PHEX. Insuficiência de Crescimento.
Abstract
INTRODUCTION: X-linked hypophosphatemic rickets is a rare osteometabolic disease caused by mutations in the PHEX gene (Neutral Phosphate Regulatory Endopeptidase), located on chromosome Xp22.11. Mutations in this gene lead to increased levels of fibroblast growth factor 23 (FGF-23), impairing phosphate absorption in renal tubular cells and intestinal epithelium.
CASE REPORTS: Case 1 Female patient, 11 years old, referred for investigation of weight and height deficit associated with bone deformity of the lower limb in varus; radiographic examinations were compatible with metaphyseal alterations suggestive of rickets.
CASE 2 Male patient, 8 years old, brother of case 1, referred to evaluate frequent falls and knee pain when walking, presented radiographic findings compatible with dolichocephaly, arching of the lower limbs in varus and metaphyseal dysplasia. Biochemical investigation of both was compatible with hypophosphatemic rickets, later genetically confirmed with identification of a pathogenic mutation in the PHEX gene.
DISCUSSION: X-linked hypophosphatemic rickets is one of the main forms of genetically inherited rickets. Affected patients usually manifest symptoms even in childhood, such as delay in weight-status growth, abnormal bone mineralization, dental alterations, craniosynostosis, in addition to osteomalacia and bone deformities in the lower limbs. Given the therapeutic advances in this area, it is essential that pediatricians recognize the early signs of this disease, so that not only is correct and accurate clinical management possible, but also that family genetic counseling is carried out.
Keywords: Rickets. Rickets, Hypophosphatemic. Familial Hypophosphatemic Rickets. PHEX Phosphate Regulating Neutral Endopeptidase. Failure to Thrive.
INTRODUÇÃO
O raquitismo é considerado uma das principais doenças de subnotificação mundial, sendo caracterizado por deformidades ósseas e falhas no crescimento.1 No longo prazo, essa doença causa baixa de massa óssea, o que configura fator de risco para fraturas osteoporóticas.
A principal causa de raquitismo é nutricional, apresentando um aumento de sua prevalência globalmente, inclusive em países desenvolvidos. 2,3 Sua principal etiologia é déficit de cálcio e vitamina D.4
O raquitismo também pode ser do tipo hipofosfatêmico, que é uma doença caracterizada por hipofosfatemia e redução da reabsorção renal de fosfato. Além disso, há uma concentração sérica normal de 25-hidroxivitamina D e um calcitriol sérico inapropriadamente normal. Os principais sinais da doença são o arqueamento progressivo dos membros inferiores, somado à redução da velocidade de crescimento após o início da deambulação.5
O raquitismo hipofosfatêmico ligado ao cromossomo X (OMIM 307800)6 constitui a forma mais comum de raquitismo hipofosfatêmico,7 sendo responsável por 80% dos casos dessa doença.8 Apresenta uma incidência de 4 a 5 a cada 100.000 nascidos vivos do sexo masculino5 e é causada por mutações no gene PHEX, localizado no cromossomo Xp22.11.6
A doença caracteriza-se por hipofosfatemia associada à hiperfosfatúria e metabolismo anormal da vitamina D. Nessa doença, o raquitismo ocorre por deficiência da mineralização da placa de crescimento, com os primeiros sintomas ocorrendo no segundo semestre de vida. O raquitismo hipofosfatêmico precisa ser precocemente diagnosticado, pois seu tratamento previne sequelas incapacitantes.9
Os pacientes portadores dessa doença apresentam raquitismo, deformidades e arqueamento principalmente de membros inferiores, dores ósseas, baixa estatura, anormalidades dentárias e cranianas, além de perda auditiva.5
O acesso ao exame molecular para o diagnóstico nem sempre é disponível no Brasil. Dessa forma, é possível suspeitar da doença através de exame físico, análise bioquímica e exames de imagem. A investigação bioquímica compreende dosagem de cálcio, fósforo sérico e urinário, cálculo da excreção renal de fósforo, além de dosagem de fosfatase alcalina, paratormônio (PTH) e vitamina D. Nos exames de imagem, as radiografias podem mostrar fraturas, arqueamentos ósseos, principalmente nos membros inferiores, deformidades e alargamento das epífises.10
O diagnóstico é usualmente confirmado por meio de concentrações reduzidas de fosfato sérico, somado a valores aumentados de fosfatúria com níveis inapropriados de calcitriol, que podem ser explicados pelo aumento do fator de crescimento de fibroblasto 23 (FGF-23) pelos osteoblastos e osteócitos,11 e/ou pela identificação do gene mutado por meio de sequenciamento genético-molecular (Figura 1).5
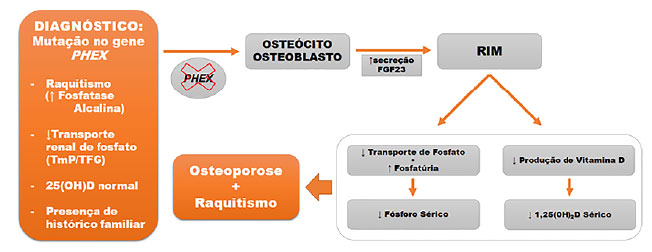
Figura 1. Fisiopatologia do raquitismo hipofosfatêmico ligado ao X. TmP: Reabsorção tubular máxima de fosfato; TFG: Taxa de filtração glomerular; 25(OH)D: Vitamina D; 1,25(OH)2D: Calcitriol (forma ativa da vitamina D); FGF23: Fator de crescimento de fibroblasto 23.
A hipofosfatemia pode não estar presente no primeiro ano de vida, o que mostra que a presença de um marcador genético para essa doença pode contribuir para o aconselhamento genético nesses casos. O tratamento é mais eficaz quando iniciado ao nascimento, portanto o diagnóstico precoce é essencial.12-14
Dessa forma, o objetivo dese relato é apresentar a investigação de três gerações afetadas pelo raquitismo hipofosfatêmico ligado ao X, no qual o diagnóstico definitivo foi estabelecido através da análise genética do gene PHEX, possibilitando uma abordagem terapêutica mais específica para a família. Dados foram obtidos através da revisão dos prontuários com a autorização do comitê de ética do hospital e dos pacientes, por assinatura do termo de consentimento livre e esclarecido.
RELATO DE CASOS
Caso 1
Paciente feminino, 12 anos, filha de um casal jovem e não consanguíneo, encaminhada, aos seis anos de idade, para investigação de deformidade progressiva em varo nos membros inferiores.
Após uma gestação sem intercorrências, nasceu de parto cesárea, com APGAR 8 e 9 no 1º e 5º minutos, respectivamente, pesando 3.460kg (percentil 50) e com comprimento de 49 cm (entre p25 e p50). Alta com 48 horas de vida sem relato de quaisquer outras queixas durante o período perinatal.
Marcos do neurodesenvolvimento revelaram-se dentro da normalidade. Apresentou, ainda, desenvolvimento cognitivo adequado com bom rendimento escolar. Paciente sempre apresentou baixo peso e crescimento em relação à idade. Com 1 ano e 4 meses, deformidade óssea em varo do membro inferior foi observada pela primeira vez, e iniciou-se acompanhamento com ortopedista sem, contudo, estabelecer etiologia da alteração em membros.
Somente aos 6 anos de idade, quando a paciente estava com marcante déficit pondero-estatural e com progressão do varismo em membros inferiores, foi encaminhada para avaliação com endocrinologista pediátrico. Nessa idade, apresentava peso de 15.75kg (<p3) e estatura de 101cm (<<<p3); presença de geno varo bilateral e de marcha com discreta báscula de quadril.
Após avaliação clínica, foram solicitados exames bioquímicos (Tabela 1) e radiográficos (Figura 2), cujos resultados foram compatíveis com suspeita clínica de raquitismo hipofosfatêmico.
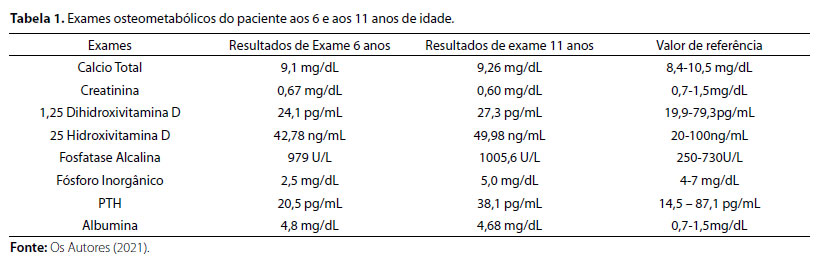
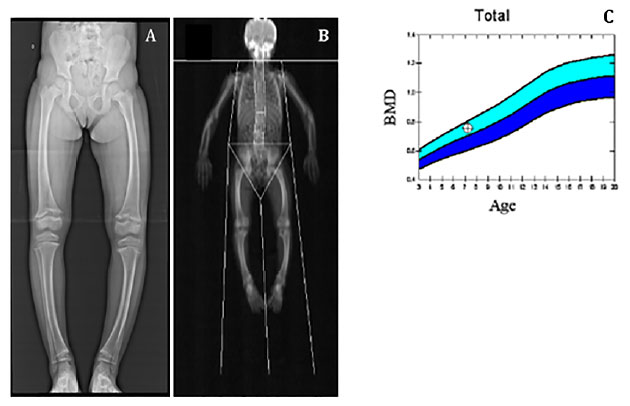
Figura 2. (A) Radiografia evidenciando discreto arqueamento de fêmur e tíbias bilaterais, alargamento e irregularidade de contornos e incipiente fragmentação, além de metáfises distais dos fêmures e proximais das tíbias esboçando aspecto em taça. (B) e (C) Densitometria óssea com massa óssea adequada para idade e sexo. Fonte: Os autores (2021).
Com o diagnóstico laboratorial, paciente iniciou reposição de fósforo (30 mg/kg/dia) e de calcitriol (0,03 mcg/kg/dia). Posteriormente, houve aumento da dose do fósforo para 40mg/kg/dia, no entanto havia muita dificuldade em se conseguir administrar a medicação na forma fracionada de quatro vezes ao dia, por dificuldade de aceitação pela criança, permanecendo com a dose inicial. Apesar de uma leve melhora na velocidade de crescimento, paciente ainda se encontrava bem abaixo da estatura esperada pela idade.
Do ponto de vista nutricional, referia baixa ingesta de leite e derivados lácteos. Não apresentava lesões odontológicas ou queixas de dor óssea.
Aos dez anos, realizou sequenciamento genético-molecular para genes envolvidos no fenótipo de raquitismo hipofosfatêmico, sendo evidenciada a presença de deleção dos exons 10 e 11 do gene PHEX, confirmando o diagnóstico bioquímico prévio de raquitismo hipofosfatêmico ligado ao X. Nessa ocasião, foram testados outros membros da família com sintomas clínicos compatíveis com raquitismo, corroborando o mecanismo de herança da enfermidade (Figura 3).
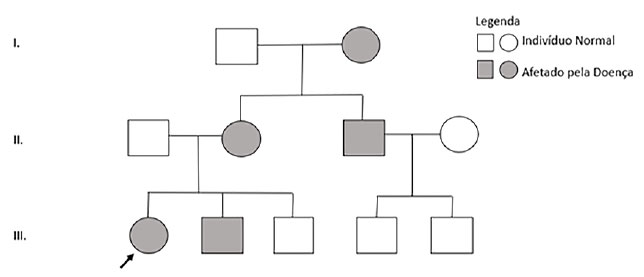
Figura 3. Heredograma familiar. Fonte: Os autores (2021).
Curiosamente, genitora da paciente apenas apesentou sintomas sugestivos de raquitismo em sua primeira gestação, apesar de a avó materna já ter um diagnóstico bioquímico compatível com raquitismo hipofosfatêmico feito há 30 anos. Tio da paciente também foi diagnosticado molecularmente e demonstrava alguns sintomas de raquitismo hipofosfatêmico, como abscessos dentários e baixa estatura.
Paciente permanece em acompanhamento clínico, mantendo reposição de fósforo (ainda que de modo irregular) e de calcitriol, contudo genitora refere que, a despeito das terapias vigentes, criança vem se queixando de piora na marcha, com acentuação do encurvamento dos membros inferiores. Não houve sinais de nefrocalcinose em exame de ultrassom renal feito aos 11 anos de idade.
Exame físico atual, aos 12 anos de idade, apresentou peso 29.3kg (<p3) e estatura 127,6cm (<<<p3), frontal amplo; face simétrica; motricidade ocular preservada; palato ogival; leve hipotonia axial; reflexos osteotendíneos normoativos; força muscular preservada; eumétrica e eudiadococinética; reflexo cutâneo-plantar em flexão bilateralmente; varismo acentuado em membros inferiores com marcha "gingada".
Caso 2
Paciente masculino, 8 anos, filho de casal jovem e não consanguíneo foi encaminhado para avaliação com endocrinologista aos 3 anos de idade, por apresentar irmã em investigação de raquitismo hipofosfatêmico e deformidade progressiva em varo nos membros inferiores.
Após uma gestação sem intercorrências, nasceu de parto cesárea, com APGAR 8 e 9 no 1º e 5º minutos, respectivamente, pesando 3.450 kg (percentil 50) e com comprimento de 47 cm (entre p3 e p10). Alta com 48 horas de vida sem relato de quaisquer outras queixas durante o período perinatal.
Marcos do neurodesenvolvimento revelaram-se dentro da normalidade. Apresentou, ainda, desenvolvimento cognitivo adequado com bom rendimento escolar. A partir de 2 anos de idade, contudo, passou a cursar com quedas frequentes e dores nos membros inferiores, em particular nos joelhos, quando caminhava distâncias um pouco maiores. Até essa idade, genitora não havia observado alterações osteoarticulares no filho, mas com a persistência dos episódios de queda, relatou surgimento progressivo de deformidade em varo de membros inferiores.
Concomitantemente, em consultas de puericultura, paciente apresentava curva de crescimento e peso sempre abaixo do esperado para idade, apesar de não haver queixas de dificuldade alimentar ou outras comorbidades.
Em virtude do quadro de raquitismo em investigação em irmã, foi encaminhado para avaliação em conjunto. Ao exame clínico, com 1 ano e 5 meses de idade, apresentava 12.8kg (~p10), 85.5cm (<<<p3) perímetro cefálico 53cm (p97); aparente macrocefalia (escafocefalia); joelhos em geno varo bilateral, sem sinais de artrite em articulações, além de marcha com báscula de quadril.
Após avaliação clínica, foram solicitados exames bioquímicos (Tabela 2) e radiográficos (Figura 4), cujos resultados foram compatíveis com suspeita clínica de raquitismo hipofosfatêmico. Estudo genético-molecular confirmou presença da deleção dos exons 10 e 11 do gene PHEX também presente na irmã do caso 2, confirmando-se o diagnóstico bioquímico prévio de raquitismo hipofosfatêmico ligado ao X.
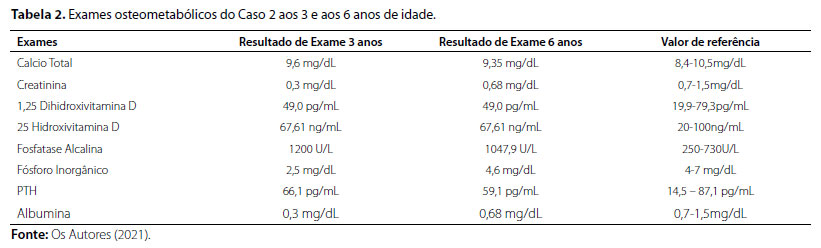
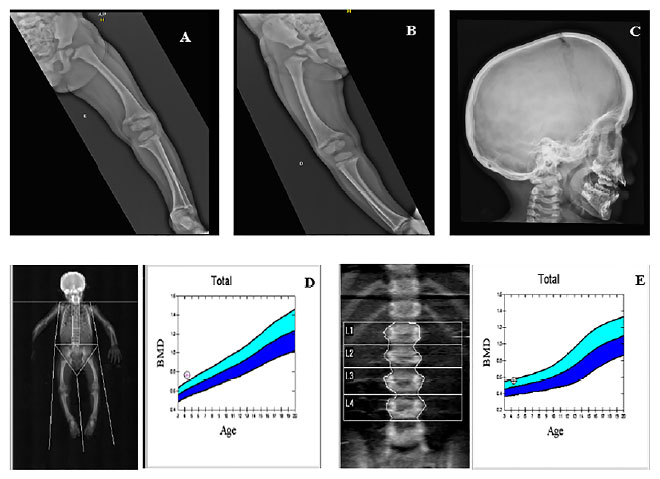
Figura 4. (A) e (B) Radiografias de ossos longos evidenciando textura óssea preservada, discreto arqueamento de fêmur e tíbia bilaterais; alargamento, irregularidade de contornos e incipiente fragmentação das metáfises distais de fêmures e tíbias, esboçando aspecto em taça. (C) Radiografia de Crânio apresentando estruturas ósseas íntegras, ausência de calcificações patológicas intracranianas, sela túrcica normal e presença de sinais de escafocefalia. (D) e (E) Densitometria óssea evidenciando aumento de massa óssea para idade e sexo. Fonte: Os autores (2021).
Paciente iniciou acompanhamento com setor de endocrinologia pediátrica e reposição de calcitriol (0,03 mcg/kg/dia) e fósforo (30 mg/kg/dia). Em função do achado de escafocefalia ao exame físico, paciente foi submetido à ressonância magnética (RNM) de encéfalo, cujo resultado foi compatível com crânio dolicocefálico com dimensões discretamente reduzidas e discreto rebaixamento das tonsilas cerebelares, sugestivo de malformação de Arnold-Chiari tipo I.
O seguimento ambulatorial, contudo, é feito de modo irregular. Houve episódios de diarreia após uso do fosfato, dificultando adesão à terapia. Concomitantemente, paciente passou a cursar com infecções de repetição (faringite), fazendo uso constante de antibioticoterapia. Diferentemente da irmã, problemas odontológicos tornaram-se recorrentes nele, com necessidade de tratamento de endodontia em toda a arcada dentária. Piora do arqueamento das pernas foi observado no período, com queixa de dor em membros inferiores, em particular pela manhã ao acordar e após realizar esforço mais intenso. Novos exames radiográficos evidenciaram progressão das lesões metafisárias, embora não houvesse sinais de nefrocalcinose em ultrassonografia renal.
Exame físico atual, aos 9 anos e 3 meses de idade, revelou: peso 22.1kg (<p3) e estatura 111,5cm (<<<p3); frontal amplo; macrocefalia com dolicocefalia; face simétrica; motricidade ocular preservada; palato ogival; reflexos osteotendíneos normoativos; força muscular preservada; eumétrico e eudiadococinético; reflexo cutâneo-plantar em flexão bilateralmente; "levantar miopático"; varismo acentuado em membros inferiores com marcha "gingada".
DISCUSSÃO
O raquitismo hipofosfatêmico familiar é principalmente devido à perda renal de fosfato; ligado ao cromossomo X (XLH), é o mais comum entre os demais tipos de raquitismo. A incidência estimada é de 1: 20.000, causada pela mutação no gene regulador de fosfato com homologia a endopeptidases (PHEX) localizado no cromossomo X.1,2 É herdado como dominante ligado ao X. O defeito fisiológico clássico em XLH é a reabsorção tubular renal proximal prejudicada de fosfato.3
A apresentação clínica em crianças com essa patologia consiste em baixa estatura, geno varo ou valgo, alargamento da metáfise, rosário raquítico e bossa frontal. Há aumento da frequência de cárie dentária ou abscessos perirradiculares e dores e rigidez óssea, muscular e articular. O arqueamento dos membros inferiores pode ser observado ao início da deambulação, ainda no primeiro ano de vida. As manifestações clínicas variam em gravidade, de assintomático à grave, de início precoce a casos com apresentação tardia (late-onset). As principais manifestações da XLH estão resumidas na Figura 5.
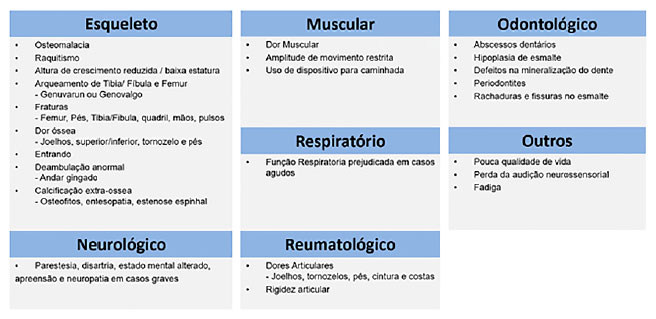
Figura 5. Principais manifestações de XLH por sistema acometido. Fonte: Os autores (2021).
Casos 1 e 2 apresentaram a maioria das manifestações descritas, embora haja algumas diferenças clínicas: dolicocefalia, abscessos dentários e sinais mais evidentes de miopatia estavam mais evidentes no caso 2, o que pode ser explicado por ser uma doença ligado ao X, portanto mais grave usualmente em pacientes do sexo masculino.
Embora pacientes do sexo feminino possam também ser igualmente afetadas em gravidade como pacientes do sexo masculino, é difícil prever o fenótipo; e mesmo dentro de uma família, mulheres portadoras da mutação no gene PHEX podem ter evoluções clínicas diferentes, o que pode ser corroborado pela mãe e avó do caso 1, que são bem menos sintomáticas que essa paciente. Isso usualmente reflete o processo de inativação randômica do cromossomo X em mulheres ("hipótese de Lyon"), ou seja, em cada paciente do sexo feminino, pode haver manifestações mais leves ou mais graves, a depender da proporção do cromossomo X que carrega a mutação no PHEX, esteja ativo ou inativo.
Como parte da investigação de qualquer paciente com suspeita de raquitismo, é fundamental que sejam realizados testes bioquímicos direcionados para essa hipótese, em particular com dosagem de fosfatase alcalina, fosforo e PTH entre outros exames (Figura 6). No entanto, a realização do teste genético confirma o diagnóstico bioquímico e auxilia não só no aconselhamento genético mas também na orientação terapêutica em alguns casos, visto que recentemente terapias específicas focadas no FGF23 foram desenvolvidas.
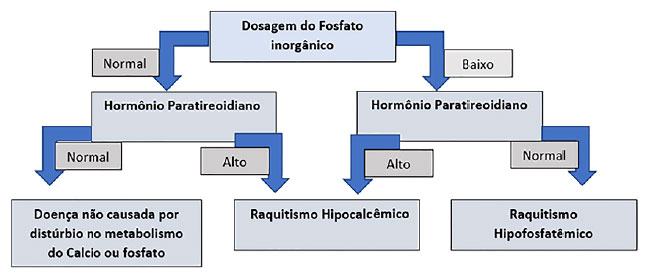
Figura 6. Algoritmo bioquímico de investigação de raquitismo. Fonte: Os autores (2021).
PHEX envolve a regulação negativa de FGF23. O FGF23 inibe a produção renal de 1,25-dihidroxivitamina D3 e a reabsorção de fosfato pela redução da 1-α-hidroxilase no túbulo proximal renal.16 Portanto, o tratamento médico da XLH inclui suplementação oral de fosfato e análogos ativos da vitamina D (alfacalcidol ou calcitriol).17
O objetivo da medicação oral, entretanto, não é normalizar o nível de fosfato sérico, mas melhorar os sintomas clínicos, como dores nos ossos e baixa estatura. Se os pacientes pediátricos assintomáticos não receberem tratamento, é provável que seus sintomas clínicos comecem antes de 1 ano de idade. Em crianças sintomáticas, a deficiência de crescimento e as anormalidades esqueléticas tendem a ser mais agravadas.11 O tratamento é previsto para crianças e jovens ainda em fase de crescimento, sendo menos claro se há benefícios para tratamento de pacientes adultos em que as placas de crescimento já estão fechadas.
O monitoramento bioquímico deve ser realizado em intervalos de 3 em 3 meses, para evitar complicações como hipercalcemia, hipercalciúria ou hiperparatireoidismo.7 O melhor biomarcador para a consolidação óssea é a atividade da fosfatase alcalina sérica, que deve diminuir com o tratamento, sugerindo uma consolidação óssea ideal.7 Nos pacientes relatados, embora o caso 2 tenha apresentado discreta diminuição dos níveis da fosfatase alcalina, paciente 1, ao contrário, evidenciou aumento da fosfatase alcalina, apontando a necessidade de ajustes terapêuticos, reforçando a importância do compliance dos pacientes no seguimento clínico de XLH, o que nem sempre é possível por uma variedade de causas, especialmente em pacientes adultos.
Os níveis de PTH devem ser medidos rotineiramente, para identificar hiperparatireoidismo secundário, que pode ser corrigido pelo aumento da dose de calcitriol ou pela redução da dose de fosfato.7 Nos casos descritos neste artigo, não houve alteração dos níveis de PTH ao longo do tratamento.
A avaliação radiológica deve ser feita para excluir o arqueamento fisiológico e a displasia óssea. Após o tratamento, a avaliação radiológica deve ser repetida para a avaliação da cicatrização do raquitismo, visando avaliar as deformidades esqueléticas e quando se considera o tratamento cirúrgico. A nefrocalcinose é uma complicação do tratamento relacionada à dose do medicamento utilizado. A ultrassonografia renal é sugerida em intervalos de 2 a 5 anos após o início do tratamento, para detectar nefrocalcinose.15 Ambos os pacientes foram avaliados por ultrassom e nenhum sinal de nefrocalcinose foi detectada.
Em adultos assintomáticos, como a genitora dos pacientes, a necessidade de tratamento médico não é óbvia porque não há evidências de benefícios clínicos ou resultados em longo prazo. Além disso, o tratamento tem efeitos colaterais, como calcificação renal e disfunção da glândula tireoide. Os casos graves requerem cirurgia, como osteotomia ou epifisiodese, porque a maioria dos pacientes sintomáticos pode receber parcialmente tratamento médico. Assim, as indicações para tratamento médico incluem sobretudo aqueles em pacientes pediátricos, adultos sintomáticos e pacientes que precisam de grandes quantidades de cálcio e fósforo, como mulheres grávidas e pacientes de cirurgia ortopédica perioperatória.
O hormônio do crescimento (rhGH) foi tentado como uma terapia adjuvante em XLHR e mostrou ter uma melhora no crescimento linear e um aumento transitório no fosfato sérico e uma diminuição transitória na excreção de fosfato urinário quando tratado com rhGH. O aumento da atividade da fosfatase alcalina sérica evidencia piora das deformidades nas pernas e da desproporção corporal, conforme relato após terapia com hormônio de crescimento.
A cirurgia durante a infância deve ser evitada. A intervenção cirúrgica deve ser considerada se o arqueamento for grave ou a torção tibial não melhorar com o tratamento médico. As osteotomias corretivas geralmente não são realizadas em crianças com menos de 6 anos de idade, uma vez que a terapia médica costuma melhorar as deformidades do arco nessa faixa etária. As osteotomias são geralmente adiadas até que o crescimento tenha quase cessado, mas deformidades graves podem exigir terapia mais precoce.
Infelizmente, mesmo a terapia com calcitriol e reposição de fósforo é muitas vezes mal sucedida, o que pode ter sido o caso dos pacientes descritos. Ademais, há diversos eventos adversos significativos associados a esse tratamento, incluindo hiperparatireoidismo secundário ou terciário, nefrocalcinose e complicações cardiovasculares, incluindo hipertensão arterial, decorrentes do hiperparatireoidismo. Além disso, esse tratamento paradoxalmente estimula a atividade do FGF23, aumentando ainda mais a perda de fosfato renal, resultando em um efeito rebote, limitando a efetividade dessa terapia. Em vista disso, uma nova possibilidade terapêutica baseada na inibição da atividade do FGF23 vem se tornando uma opção mais específica e sem incorrer nos efeitos adversos previamente observados.
Indicada para pacientes com raquitismos hipofosfatêmicos causados pelo excesso de FGF23, o burosumabe (Crysvita®) é um anticorpo monoclonal que se liga e inibe a atividade do fator de crescimento de fibroblastos 23.18 Ao inibir o FGF23, espera-se que o burosumabe aumente a reabsorção de fosfato pelo rim e, através da produção de vitamina D, melhore a absorção intestinal de cálcio e fosfato.19 Resultados clínicos de diversos estudos realizados nos últimos anos apontam para bons resultados dessa terapia,20,21 e o mesmo já foi aprovado em diversos países por suas respectivas agências reguladoras, inclusive no Brasil pela Anvisa.
Em conclusão, pacientes com baixa estatura, em particular com presença de achados ósseos como geno valgo/geno varo, devem ser investigados para raquitismo a partir de testes bioquímicos iniciais, confirmando-se posteriormente com análise genético-molecular. A investigação genética não só permite a elucidação da etiologia da doença, mas também pode auxiliar na orientação de terapia mais específica para os pacientes e, claro, possibilitar o aconselhamento genético familiar.
REFERÊNCIAS
1. Elder CJ, Bishop NJ. Rickets. The Lancet. maio de 2014;383(9929):1665-76.
2. Prentice A. Nutritional rickets around the world. The Journal of Steroid Biochemistry and Molecular Biology. julho de 2013;136:201-6.
3. Cashman KD, Dowling KG, Skrabáková Z, Gonzalez-Gross M, Valtueña J, De Henauw S, et al. Vitamin D deficiency in Europe: pandemic? The American Journal of Clinical Nutrition. 1o de abril de 2016;103(4):1033-44.
4. Uday S, Högler W. Nutritional Rickets and Osteomalacia in the Twenty-first Century: Revised Concepts, Public Health, and Prevention Strategies. Curr Osteoporos Rep. agosto de 2017;15(4):293-302.
5. Ruppe MD. X-Linked Hypophosphatemia. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Stephens K, et al., organizadores. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 2017 [citado 19 de junho de 2020]. Disponível em: http://www.ncbi.nlm.nih.gov/books/NBK83985/
6. OMIM 307800. HYPOPHOSPHATEMIC RICKETS, X-LINKED DOMINANT; XLHR [Internet]. [citado 17 de junho de 2020]. Disponível em: https://www.omim.org/entry/307800
7. Emma F, Cappa M, Antoniazzi F, Bianchi ML, Chiodini I, Eller Vainicher C, et al. X-linked hypophosphatemic rickets: an Italian experts' opinion survey. Ital J Pediatr. dezembro de 2019;45(1):67.
8. handran M, Chng CL, Zhao Y, Bee YM, Phua LY, Clarke BL. Novel PHEX Gene Mutation Associated with X Linked Hypophosphatemic Rickets. Nephron Physiol. 2010;116(3):p17-21.
9. Bitzan M, Goodyer PR. Hypophosphatemic Rickets. Pediatric Clinics of North America. fevereiro de 2019;66(1):179-207.
10. Maia ML de A, Abreu ALS, Nogueira PCK, Val MLDM do, Carvalhaes JT de A, Andrade MC de. RAQUITISMO HIPOFOSFATÊMICO: RELATO DE CASO. Rev paul pediatr. 29 de março de 2018;36(2):242-7.
11. Oliveira RB de, Moysés RMA. FGF-23: estado da arte. J Bras Nefrol. setembro de 2010;32(3):323-31.
12. Nunes AB, Lazaretti-Castro M. Raquitismo hipofosfatêmico: da clínica à genética molecular. Arq Bras Endocrinol Metab. abril de 2000;44(2):125-32.
13. Lecoq A-L, Brandi ML, Linglart A, Kamenický P. Management of X-linked hypophosphatemia in adults. Metabolism. fevereiro de 2020;103:154049.
14. López-Romero LC, Broseta JJ, Guillén Olmos E, Devesa-Such RJ, Hernández-Jaras J. Raquitismo hipofosfatémico ligado al cromosoma X: diagnóstico en la edad adulta y forma paucisintomática. Reumatología Clínica. novembro de 2019;S1699258X19301214.
15. Alenazi B, Molla MAM, Alshaya A, Saleh M. Raquitismo hipofosfatêmico ligado ao X (mutação PHEX): Um relato de caso e revisão da literatura. Sudan J Paediatr. 2017; 17 (1): 61-65.
16. Yang M, Kim J, Yang A, Jang J, Jeon TY, Cho SY, Jin DK. Uma nova mutação em mosaico em PHEX em um paciente coreano com raquitismo hipofosfatêmico. Ann Pediatr Endocrinol Metab. 2018 dez; 23 (4 ): 229-234.
17. Imel EA, Glorieux FH, Whyte MP, Munns CF, Ward LM, Nilsson O, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, activecontrolled, open-label, phase 3 trial. Lancet.2019;393(10189):2416-27.
18. Carpenter TO, Whyte MP, Imel EA, Boot AM, Högler W, Linglart A, et al. Burosumab therapy in children with X-linked hypophosphatemia. N Engl J Med. 2018;378(21):1987-98.
19. Whyte M, Carpenter T, Gottesman G, Mao M, Skrinar A, San Martin J, et al. Efficacy and safety of burosumab in children aged 1-4 years with X-linked hypophosphataemia: a multicentre, openlabel, phase 2 trial. Lancet Diabetes Endocrinol [Internet]. 2019;7(3):189-99.
20. Insogna KL, Briot K, Imel EA, Kamenický P, Ruppe MD, Portale AA, et al. A Randomized, DoubleBlind, PlaceboControlled, Phase 3 Trial Evaluating the Efficacy of Burosumab, an Anti-FGF23 Antibody, in Adults With X-Linked Hypophosphatemia: Week 24 Primary Analysis. J Bone Miner Res. 2018;33(8):1383-93. 61.
21. Insogna KL, Rauch F, Kamenický P, Ito N, Kubota T, Nakamura A, et al. Burosumab Improved Histomorphometric Measures of Osteomalacia in Adults with X-Linked Hypophosphatemia: A Phase 3, Single-Arm, International Trial. J Bone Miner Res. 2019;34(12):2183-91.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()