Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(1) - Março 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Histiocitose de células de langerhans: Relato de caso com ênfase no diagnóstico diferencial
Langerhans cell histiocytosis: Case report with emphasis in differential diagnosis
Ronaldo Almeida Lidório-Júnior1; Maria Giovana Queiroz de-Lima1; Lúcia Alves da-Rocha2; Alessandra Encarnação de-Morais1; Juan Eduardo Rios Rodriguez1; Pedro Fernandes Santos1; Lucas de Moraes Martins Pereira1; Natalia Leal Epifânio3
DOI:10.31365/issn.2595-1769.v21i1p36-39
1. Universidade Federal do Amazonas, Departamento de Pediatria - Manaus - Amazonas - Brasil
2. Fundação de Medicina Tropical Doutor Heitor Vieira Dourado, Núcleo de Investigação em Arboviroses, Roboviroses e Viroses Emergentes do Amazonas/ FMT-HVD - Manaus - Amazonas - Brasil
3. Fundação de Hematologia e Hemoterapia do Amazonas - HEMOAM, Departamento de Ensino e Pesquisa - Manaus - Amazonas - Brasil
Endereço para correspondência:
Recebido em: 25/08/2020
Aprovado em: 15/02/2021
Instituição: Universidade Federal do Amazonas, Departamento de Pediatria - Manaus - Amazonas - Brasil
Resumo
INTRODUÇÃO: Histiocitose de células de Langerhans é uma neoplasia inflamatória causada pela multiplicação de células aberrantes do sistema mononuclear fagocítico. Trata-se da desordem histiocitária mais comum, afetando cerca de 5 crianças por milhão, na faixa etária de 0 a 15 anos, similar à frequência do linfoma Hodgkin.
OBJETIVO: Relatar um caso de histiocitose de células de Langerhans de alto risco, destacando a importância do diagnóstico diferencial em casos de linfadenopatia e as dificuldades geográficas do Amazonas para seguimento adequado do paciente.
DESCRIÇÃO DO CASO:Adolescente de 13 anos, do interior do estado do Amazonas, apresentou linfadenopatia em região cervical, axilar e supra-clavicular. Foi realizado tratamento para tuberculose ganglionar, mas as lesões pioraram com o passar do tempo, indicando a necessidade de realização de biópsia. Foi, então, constatada histiocitose de células de Langerhans, confirmada pela imuno-histoquímica. No momento, o paciente se encontra em tratamento para a condição, com boa resposta terapêutica a despeito das dificuldades geográficas e socioeconômicas.
DISCUSSÃO: O espectro clínico vasto da histiocitose dificulta seu diagnóstico precoce. O paciente apresentou acometimento linfonodal, raro para essa doença e fato que suscitou o diagnóstico diferencial com tuberculose ganglionar, a primeira hipótese sugerida para a criança. Portanto, é de suma importância considerar a histiocitose no grupo de doenças que cursam com linfadenopatia cervical. Além disso, o caso suscita a discussão em torno de questões geográficas e como as mesmas podem interferir para diagnóstico e tratamento adequados.
Palavras-chave: Histiocitose; Histiocitose de Células de Langerhans; Linfadenopatia.
Abstract
INTRODUCTION: Langerhans cell histiocytosis is an inflammatory neoplasm caused by the multiplication of aberrant cells in the phagocytic mononuclear system. It is the most common histiocytic disorder, affecting about 5 children per million, aged 0 to 15 years, similar to the frequency of Hodgkins lymphoma.
OBJECTIVE: To report a case of high-risk Langerhans cell histiocytosis, highlighting the importance of differential diagnosis in cases of lymphadenopathy and the geographical difficulties of Amazon State for adequate patient follow-up.
CASE DESCRIPTION: A 13-year-old teenager, from the interior of the Amazon State, presented lymphadenopathy in the cervical, axillary and supra-clavicular area. Treatment for ganglionic tuberculosis was performed, but the lesions worsened over time, indicating the need for biopsy. Then, Langerhans cell histiocytosis was confirmed, confirmed by immunohistochemistry. At the moment, the patient is undergoing treatment for this condition, with a good therapeutic response despite the geographical and socioeconomic difficulties.
DISCUSSION: The wide clinical spectrum of histiocytosis hinders its early diagnosis. The patient presented with lymph node involvement, rare for this disease and a fact that led to the differential diagnosis with ganglionic tuberculosis, the first hypothesis suggested for the child. Therefore, it is extremely important to consider histiocytosis in the group of diseases that present cervical lymphadenopathy. In addition, the case raises the discussion around geographic issues and how they can interfere for proper diagnosis and treatment.
Keywords: Histiocytosis; Histiocytosis, Langerhans-Cell; Lymphadenopathy.
INTRODUÇÃO
Histiocitose de células de Langerhans é uma neoplasia inflamatória causada pela multiplicação de células aberrantes do sistema mononuclear fagocítico1. Tais células são histiócitos, ou seja, células teciduais fagocitárias, derivadas de precursores mieloides imaturos, que expressam CD 207 (langerina) e CD1a e contêm grânulos de Birbeck. Há, portanto, forte semelhança com a células dendríticas localizadas na epiderme e na mucosa, as células de Langerhans2.
A doença ocorre quando tais células aberrantes se proliferam, gerando lesões granulomatosas que podem surgir em qualquer órgão ou sistema, tendo afinidade por osso, pele, pulmões e hipófise3. O espectro clínico da doença é vasto, variando de lesões cutâneas indolentes de remissão espontânea até quadro graves de comprometimento multissistêmico, em especial quando do acometimento de órgãos de alto risco, que incluem fígado, baço e medula óssea3.
Trata-se da desordem histiocitária mais comum, afetando cerca de cinco crianças por milhão, na faixa etária de 0-15 anos, similar à frequência do linfoma Hodgkin4. A maior incidência se dá antes de um ano de idade, e a média de idade do diagnóstico é de 3,5 anos4. Apesar de predominante na faixa etária pediátrica, casos em adultos e idosos também são descritos5.
A histiocitose de células de Langerhans era clinicamente dividida em fenótipos característicos, que incluíam doença de Letterer-Siwe; granuloma eosinofílico; doença de Hand-Schuller-Christian; e doença de Hashimoto-Pritzke6. Entretanto, atualmente, esta forma de classificação mostrou-se insuficiente, e a classificação mais importante da doença envolve a extensão (mono ou multissistêmica) e o acometimento de órgão de risco, quadro associado a uma menor sobrevida6.
A maioria dos pacientes apresenta o envolvimento de um único sistema (70%), enquanto dentre os acometidos por uma doença multissistêmica, cerca de 50% têm acometimento de órgão de risco7. Os órgãos mais afetados pela doença são ossos (80%), pele (33%), glândula hipófise (25%), fígado (15%), baço (15%), sistema hematopoiético (15%), pulmões (15%), linfonodos (5-10%) e sistema nervoso central (2-4%)8.
DESCRIÇÃO DO CASO
J.N.D, 13 anos, masculino, pardo, procedente de Borba, interior do estado do Amazonas, procurou, em outubro de 2017, hospital referência em doenças tropicais referindo quadro de tosse, febre e perda ponderal não mensurada há seis meses, associado a linfadenopatia cervical, axilar e supraclavicular bilateralmente. Nesse período, realizou tomografia computadorizada (TC) cervical, que detectou linfonodos aumentados à direita, além de TC de tórax, cuja conclusão foi de infiltrado micronodular ramificado (árvore de brotamento) bilateral com pequeno derrame pleural em hemitórax direito (sugestivo de exsudato) e linfonodos aumentados de volume nas axilas e no hilo direito. Dentro do contexto, considerou-se a hipótese diagnóstica de tuberculose pulmonar e ganglionar, e apesar do teste rápido para tuberculose negativo, foi iniciado o tratamento.
Durante o tratamento, o paciente obteve melhora do quadro respiratório, mas houve aumento de linfadenopatia axilar bilateral, com surgimento de áreas de necrose e drenagem de secreção fétida amarelada à direita. As lesões tinham aspecto vegetativo e eram muito dolorosas, fazendo-o buscar serviço de referência dermatológica em março de 2018. Foi solicitado exame anatomopatológico da lesão linfonodal (figura 1), cujo resultado revelou processo linfogranulomatoso atípico, com pesquisa para BAAR negativa. Exame micológico direto e cultura também foram realizados, ambos negativos. Nova TC de tórax mostrou resolução do infiltrado micronodular, além de redução do pequeno derrame pleural em hemitórax direito e piora de linfonodomegalias. Exames sorológicos requeridos no período (VHB/VHC/HIV/HTLV) foram negativos.

Figura 1. Laudo de Exame Anátomo Patológico
Em abril de 2018, foi admitido em um pronto socorro infantil de Manaus para avaliação cirúrgica das lesões infra-axilares, sem outras alterações ao exame físico. O teste rápido molecular (PCR) não detectou DNA para M. tuberculosis. Devido à dúvida diagnóstica e quadro arrastado com piora das lesões, optou-se por abordar cirurgicamente o local, através da exérese de lesão de pele abcedada e biópsia de gânglio axilar esquerdo.
Novo exame anatomopatológico de gânglio axilar esquerdo revelou arquitetura subvertida às custas de células gigantes multinucleadas, algumas fagocitando linfócitos (linfofagocitose), de permeio a aglomerados de granulócitos neutrófilos, raros granulócitos eosinófilos, histiócitos espumosos e pigmentos de melanina. A conclusão foi de histiocitose de células de Langerhans, confirmada com estudo imunohistoquímico (figura 2), que demonstrou expressão de S100, CD1A, CD2017 e Ki-67.
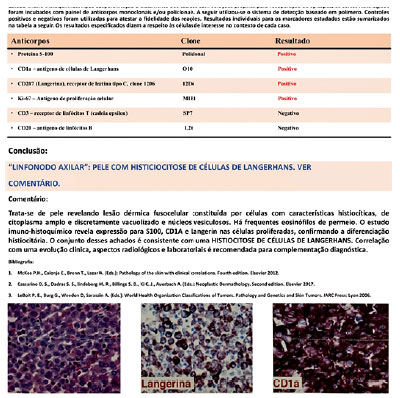
Figura 2. Estudo Imuno-histoquímico
Após, foi referenciado ao serviço especializado de Hematologia. O rastreio sistêmico da doença não revelou alterações laboratoriais ou de imagem em tórax, abdome e ossos longos, exceto pela disfunção hepática, evidenciada no aumento da fosfatase alcalina. Pelo fato de ser uma doença multissistêmica, foi realizado mielograma, cujo resultado foi de hipoplasia eritroide, sem a possibilidade de realizar imuno-histoquímica no serviço. Os exames laboratoriais indicaram presença de disfunção hepática (aumento da fosfatase alcalina), dessa forma o paciente foi classificado como alto risco e foi proposto o tratamento com corticoterapia e quimioterapia com Vimblastina. Entretanto, o paciente retornou ao seu município, tendo iniciado o tratamento apenas em janeiro de 2019, época em que regressou a Manaus.
Até setembro de 2019, o paciente estava na fase de manutenção, com Mercaptopurina em uso contínuo, Metotrexate semanal e quimioterapia com Vimblastina a cada três semanas, mas interrompeu o tratamento devido às dificuldades logísticas na permanência em Manaus.
DISCUSSÃO
Histiociotose de células de Langerhans apresenta espectro clínico vasto, fato que dificulta um diagnóstico precoce. Estudo institucional demonstrou uma média de três meses para o diagnóstico de lesões cutâneas em um centro de referência, mas deixou claro que alguns casos chegaram até cinco anos desde a apresentação até o diagnóstico definitivo7. O caso em tela evidencia nitidamente a dificuldade diagnóstica, dada a diversidade de apresentações da doença, somada às barreiras geográficas do Amazonas, com restrição de deslocamentos muitas vezes ao transporte fluvial, e a escassez de acesso a serviços especializados em contexto de interior.
O paciente apresentou acometimento linfonodal, raro para essa doença8 e fato que suscitou o diagnóstico diferencial com tuberculose ganglionar, a primeira hipótese sugerida para a criança. Portanto, é de suma importância considerar a histiocitose no grupo de doenças que cursam com linfadenopatia cervical, entre as quais infecção orofaríngea (viral, estreptocócica e estafilocócica), infecção no couro cabeludo, linfadenite por micobactérias, infecção viral (EBV, CMV, Herpes tipo 6), doença da arranhadura do gato, toxoplasmose, doença de Kawasaki, doença da tireoide, doença de Kikuchi e doença linfoproliferativa autoimune. Além do rol de patologias que cursam com linfonodomegalia supraclavicular, tais como malignidade ou infecção no mediastino, metástase de neoplasia abdominal, linfoma e tuberculose9.
O quadro pulmonar do paciente sugeriu, em um primeiro momento, tuberculose pulmonar, devido ao infiltrado em árvore de brotamento em lobos superiores associado a síndrome consumptiva e tosse crônica. Entretanto, o achado de infiltrado micronodular de aspecto ramificado é também característico de histiocitose. Nesta, é esperado um infiltrado que combine nódulos e cistos10. Importante ressaltar que os pulmões já não são mais considerados órgãos de risco, uma vez que seu acometimento não mostrou ser um fator prognóstico significativo para sobrevida10, apesar das controvérsias.
Importante ressaltar que o fato de acometer órgãos de risco indica resistência aos efeitos do tratamento quimioterápico, já que o mesmo é incapaz de curar mais de 50% dos pacientes pediátricos de alto risco e a chance de graves sequelas neurodegenerativas é maior11. Estudo prospectivo demonstrou que a sobrevida em cinco anos dos pacientes de alto risco foi de 84%, comparado a 99% aos de baixo risco, conforme ensaio clínico12.
O paciente em questão apresentou boa resposta ao tratamento inicial, conferindo melhor prognóstico. A resposta inicial à quimioterapia, durante as seis primeiras semanas, também é importante fator prognóstico nos pacientes de elevado risco. A sobrevida foi de 95% naqueles pacientes com boa resposta após as primeiras semanas de tratamento12.
Apesar das recentes descobertas das vias de sinalização acometidas na histiocitose de células de Langerhans, tais como a ativação de MAPK, o tratamento da doença ainda se mostra muito atrelado à quimioterapia, que apesar de ter demonstrado aumentar a sobrevida dos pacientes, ainda falha no tratamento daqueles com acometimento de alto risco.
Conclui-se, a partir deste caso clínico, que a histiocitose de células de Langerhans deve ser considerada no diagnóstico diferencial de linfadenopatias cervicais e supraclaviculares na infância, no sentido de possibilitar a abordagem adequada e o mais urgente possível a essa complexa doença. Além disso, o presente relato evoca uma discussão em torno da acessibilidade em saúde e impõe uma reflexão sobre os obstáculos enfrentados pela maioria da população do interior do estado do Amazonas em ser contemplada com assistência em saúde digna.
REFERÊNCIAS
1. Berres ML, Merad M, Allen CE. Progress in understanding the pathogenesis of Langerhans cell histiocytosis: Back to Histiocytosis X? Br J Haematol. 2015;169(1):3-13.
2. Abla O, Fraitag S, Horne A, Haroche J, Donadieu J, Requena-caballero L, et al. Review Article Revised classification of histiocytoses and neoplasms of the macrophage- dendritic cell lineages. Blood. 2016;127(22):2672-82.
3. Allen CE, Merad M, McClain KL. Langerhans-Cell Histiocytosis. N Engl J Med [Internet]. 2018;379(9):856-68. Available from: http://www.ncbi.nlm.nih.gov/pubmed/30157397%0Ahttp://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=PMC6334777
4. Guyot-Goubin A, Donadieu J, Barkaoui M, Bellec S, Thomas C, Clavel J. Descriptive epidemiology of childhood Langerhans cell histiocytosis in France, 2000-2004. Pediatr Blood Cancer [Internet]. 2008 Jul [cited 2019 Sep 27];51(1):71-5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/18260117
5. Mosqueira CB, Xavier AFDP, Tuschinski CL, Lopes Pinto CA, Cunha PR. Case for diagnosis. An Bras Dermatol. 2010;85(1):107-8.
6. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children: History, classification, pathobiology, clinical manifestations, and prognosis. J Am Acad Dermatol [Internet]. 2018;78(6):1035-44. Available from: https://doi.org/10.1016/j.jaad.2017.05.059
7. Simko SJ, Garmezy B, Abhyankar H, Lupo PJ, Chakraborty R, Lim KPH, et al. Differentiating skin-limited and multisystem langerhans cell histiocytosis. In: Journal of Pediatrics. Mosby Inc.; 2014. p. 990-6.
8. Haupt R, Minkov M, Astigarraga I, Schäfer E, Nanduri V, Jubran R, et al. Langerhans cell histiocytosis (LCH): Guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013 Feb;60(2):175-84.
9. Matos LL de, Faro Junior MP, Kanda JL, Gerardi Filho VA, Fernandes PM. Linfadenopatia cervical na infância: etiologia, diagnóstico diferencial e terapêutica. Arq Bras Ciências da Saúde. 2010;35(3).
10. Ronceray L, Pötschger U, Janka G, Gadner H, Minkov M. Pulmonary involvement in pediatric-onset multisystem langerhans cell histiocytosis: Effect on course and outcome. J Pediatr. 2012;161(1).
11. Gadner H, Minkov M, Grois N, Pötschger U, Thiem E, Aricò M, et al. Therapy prolongation improves outcome in multisystem Langerhans cell histiocytosis. file:///Users/tame/Desktop/Blood 2013; 121- 5006-14. .pdf. Blood. 2013;121(25):5006-14.
12. Helmut G, Nicole G, Milen M, Ulrike P, Elfriede T. Treatment Protocol of the Third International Study for LANGERHANSCELL HISTIOCYTOSIS. 2002;(April 2001):56. Available from: https://www.skion.nl/workspace/uploads/lchiiiprot-version2.pdf
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()