Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 21(1) - Março 2021
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Síndrome de Hurler: Desafio no Manejo Clínico e Importância do Diagnóstico Precoce
Hurley Syndrome: Clinical Management Challenge and Value of Early Diagnosis
Joyce Roncolato Bisinoto1; Stella Custodio Godinho1; Gabriela Diniz Mussi1; Laura Vagnini2; Marcela Almeida1; Debora de Cassia Tomaz3; Patricia Barros Viegas Anno3; Regina Albuquerque3; Zumira Aparecida Carneiro1; Ana Paula Andrade Hamad4; Jacqueline Fonseca5; Tainá Regina Damaceno Silveira6; Charles Marques Lourenco1,2,3
DOI:10.31365/issn.2595-1769.v21i1p20-25
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Centro Paulista de Diagnostico e Pesquisa, Genética Clínica - Ribeirão Preto - SP - Brasil
3. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
4. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - Ribeirão Preto - Please Select - Brasil
5. Laboratório DLE, Genética Bioquímica - Rio de Janeiro - RJ - Brasil
6. Centogene, Genética Molecular - Rostock - Mecklemburgo-Pomerânia Ocidental - Alemanha
Endereço para correspondência:
Recebido em: 05/03/2020
Aprovado em: 23/11/2020
Instituição: Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
Resumo
A mucopolissacaridose tipo I (MPS I) é uma doença rara que se caracteriza por deficiência da a- l-iduronidase, enzima lisossomal responsável pelo metabolismo dos glicosaminoglicanos (GAGs) de sulfato de dermatan e sulfato de heparan, causando um acumulo patológico dos mesmos no organismo. A doença é hereditária, autossômica recessiva e seus fenótipos podem variar. Seus sinais e sintomas iniciais podem ser específicos como opacidade ocular e deformidade da coluna vertebral ou podem ser confundidos com os de várias outras patologias como infecções auditivas recorrentes e até mesmo perda auditiva, hérnias, sintomas cardíacos e infecções respiratórias frequentes, dificultando o diagnóstico e, consequentemente, o tratamento, o qual, se iniciado de maneira precoce, poderia evitar o surgimento de sequelas irreversíveis, além de reduzir significativamente a progressão da doença. Relatamos caso de paciente, seis anos de idade, sexo feminino, diagnosticada com MPS I com 20 meses. Segundo sua história clínica, a mesma já apresentava atraso neuropsicomotor, hipertrofia de ventrículo esquerdo, infecções de vias aéreas de repetição, adenoidectomia e duas herniorrafias umbilical e inguinal, porém sem estabelecimento de diagnóstico de base. O diagnóstico correto, ainda que tardio, possibilitou início de terapia de reposição enzimática (TRE), melhor acompanhamento de possíveis complicações clínicas, além do aconselhamento genético da família.
Palavras-chave: Mucopolissacaridose I; Glicosaminoglicanos; Diagnóstico Precoce; Terapia de Reposição de Enzimas.
Abstract
Mucopolysaccharidosis type I (MPS I) is a rare disease characterized by deficiency of a-L-iduronidase, a lysosomal enzyme responsible for the degradation of glycosaminoglycans (GAGs) of dermatan sulfate and heparan sulfate, leading to pathological GAGs accumulation in the body. It is a hereditary autosomal recessive disorders and its phenotypes may vary. Its early signs and symptoms can be specific such as ocular opacity and spinal deformity or can mimic those from many other diseases such as recurrent ear infections and even hearing loss, herniae, heart symptoms and frequent respiratory infections, leading to difficulties in diagnosis and treatment. Early treatment can avoid the appearance of irreversible sequela and decrease significantly disease progression. Here we report a six year old female patient with a late diagnosis of MPS I when she was 20 months old. According to its clinical history, although no specific baseline diagnosis was established, she already presented at that age neuropsychomotor retardation, left ventricular hypertrophy, recurrent airway infections, adenoidectomy and two umbilical and inguinal herniorraphies. The correct diagnosis, even though late, allowed the initiation of enzyme replacement therapy (ERT), better follow-up of possible clinical complications and also family genetic counseling.
Keywords: Mucopolysaccharidosis I; Glycosaminoglycans; Early Diagnosis; Enzyme Replacement Therapy.
INTRODUÇÃO
Mucopolissacaridoses (MPSs) são doenças de armazenamento lisossômico causadas pelo acúmulo de mucopolissacarídeos (longas moléculas de açúcar), mais modernamente chamados de glicosaminoglicanos (GAGs). São doenças metabólicas raras, atualmente apresentadas em sete tipos diferentes (I, II, III, IV, VI , VII e IX).1
A mucopolissacaridose tipo I (MPS I, também conhecida como síndrome de Hurler) foi umas das primeiras formas de MPS a ser descrita.2 Trata-se de uma doença de armazenamento lisossomal com a frequência de ocorrência relatada de 0,99 a 1,99 para cada 100.000 nascidos vivos.3 Caracteriza-se por deficiência da α-l-iduronidase, enzima lisossomal responsável pelo metabolismo dos GAGs de sulfato de dermatan e sulfato de heparan causando um acúmulo patológico dos mesmos no organismo, especificamente nos lisossomos. Com o acúmulo progressivo dos GAGs, componentes essenciais da estrutura de ossos, cartilagem, pele, tendões e outros tecidos do corpo, pode advir comprometimento de diversos órgãos, particularmente o encéfalo.2
A MPS I possui caráter hereditário, autossômica recessiva, com ampla variabilidade fenotípica. Atualmente, ela pode ser classificada em MPS I-forma clássica (síndrome de Hurler), MPS intermediária (síndrome de Hurler-Scheie) e MPS I-leve (síndrome de Scheie), classificadas de acordo com a presença ou ausência de envolvimento neurocognitivo e progressão da doença que pode causar incapacidade e levar à morte.4
Muitos médicos a desconhecem e seus sinais e sintomas iniciais cursam com os de várias outras patologias, como opacidade ocular, infecções auditivas recorrentes e até mesmo perda auditiva, hérnias, deformidade da coluna vertebral, sintomas cardíacos e infecções respiratórias frequentes, dificultando o diagnóstico e, consequentemente, o tratamento, que quando precoce podem reduzir significativamente a progressão da doença.4
O tratamento atual da síndrome de Hurler compreende a terapia de reposição enzimática (TRE), que consiste na aplicação periódica de uma enzima recombinante por via intravenosa, e o transplante de células-tronco hematopoiéticas (TCTH), que deve ser realizado, preferencialmente, ainda antes dos dois anos.2 O sucesso das terapias atuais pode ser influenciado por vários fatores e deve ser monitorizado com a avaliação clínica e uso de biomarcadores (como a dosagem dos GAGs urinários).
Relatamos o caso de paciente brasileira, portadora de síndrome de Hurler, e os desafios no manejo das complicações clínicas associadas a essa enfermidade.
RELATO DE CASO
Paciente do sexo feminino, seis anos de idade, foi encaminhada para investigação de doença de depósito lisossomal. Trata-se da primeira filha de casal jovem, saudável e não consanguíneo.
Seus antecedentes gestacionais e exames pré-natais da genitora não revelaram anormalidades. Nasceu prétermo (36 semanas), de parto cesáreo com 2.900g, comprimento de 46cm, perímetro cefálico 31cm, perímetro torácico 30cm e APGAR seis no primeiro minuto e oito no quinto minuto. Após o nascimento, ficou por sete dias no centro de terapia intensiva (CTI) em decorrência de uma baixa saturação e dispneia. Após este período, obteve alta e não se observaram outras intercorrências nos primeiros meses de vida.
Segundo história clínica, os antecedentes de desenvolvimento neuropsicomotor não revelaram atraso significativo de marcos motores, visto que a paciente apresentou sustento cervical aos três meses, sentou sem apoio aos sete meses, engatinhou aos dez meses, deambulou sem apoio aos 12 meses, porém apresentava atraso de fala, falando as primeiras palavras com 20 meses. Apresentava infecções de vias aéreas de repetição, adenoidectomia, duas herniorrafias (umbilical e inguinal) e roncos noturnos com apneias frequentes quando foi examinada aos 15 meses.
Em seu exame físico inicial, observaram-se nariz em sela, lábios volumosos, cifose, escoliose, hérnia umbilical, mãos pequenas e membros inferiores arqueados, sinais clínicos que sugeriam a suspeita de uma doença de armazenamento lisossômico (figura 1).
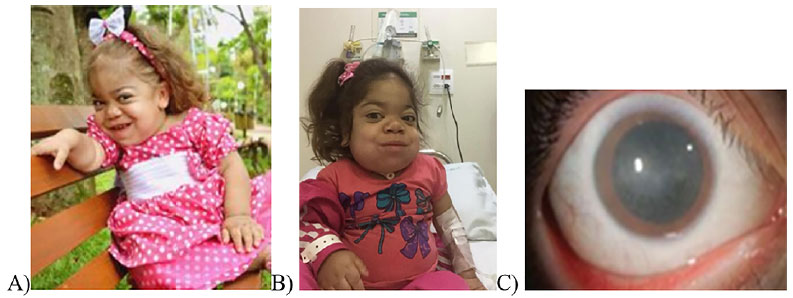
Figura 1. Sinais clínicos sugestivos de MPS I na paciente. aos 20 meses (1A) e com progressão da infiltração facial já aos cinco anos de idade em uso de traqueostomia (1B); presença de opacidade corneana (1C). Fonte: os autores (2018).
Em virtude desses achados, paciente foi encaminhada para avaliação e acompanhamento com médico geneticista. Seu exame genético-clínico revelou: estatura abaixo do percentil 3; peso entre os percentis 50 e 75; perímetro cefálico entre os percentis 75 e 97, fáceis infiltrado, dorso nasal curto, raiz nasal achatada, lábios grossos, contratura e rigidez das articulações, mãos em garra, cabelo espesso em crânio, hepatoesplenomegalia, reflexos osteotendíneos grau I em membros inferiores e superiores; ausência de sinais de compressão medular.
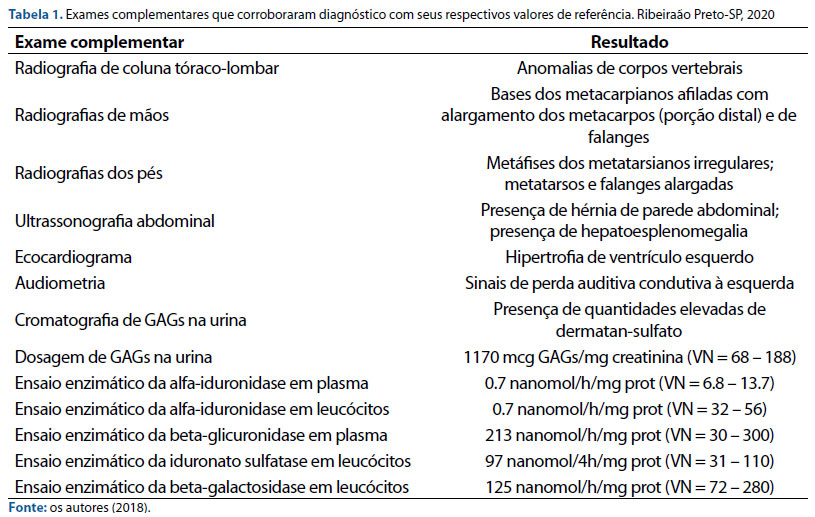
Diante da forte suspeita de uma forma de mucopolissacaridose, foram realizados exames complementares (tabela 1), os quais confirmaram o diagnóstico de MPS I quando a paciente tinha 20 meses.
Iniciou o tratamento de reposição enzimática aos 27 meses, sendo infundidos 15mg semanalmente. Após esse tratamento, observou-se aumento da estatura da paciente e redução da hepatoesplenomegalia. Aos quatro anos, foi diagnosticada com hidrocefalia, que cursava com aumento da pressão intracraniana, sendo necessária a colocação de derivação. Em decorrência de contaminação, a derivação foi retirada após 30 dias e não houve necessidade de recolocação.
Durante um exame de ressonância magnética, apresentou parada cárdio-respiratória, que é sinal de gravidade da enfermidade em pacientes com mucopolissacaridose. Foi para o CTI, onde teve mais duas paradas cardiorrespiratórias, sendo necessária intervenção cirúrgica para colocação de traqueostomia, que permanece até os dias atuais. Recentemente, evoluiu com quadro de baixa acuidade visual e em consulta oftalmológica foi observada opacidade da córnea (figura 1C), mas que não impedia refração e não foram evidenciados edema e atrofia no nervo óptico.
Aos cinco anos de idade, foi feita nova tomografia computadorizada do crânio, evidenciando alargamento das cisternas basais e suprasselar, hiperdensidade periventricular ao nível supratentorial, dilatação das cavidades ventriculares e acentuação dos sulcos corticais e fissuras silvianas bilateral na região parietal.
Atualmente, paciente encontra-se com grande déficit auditivo, com progressão há um ano e está aguardando audiometria para melhor investigação.
DISCUSSÃO
As MPSs são um grupo de doenças que se caracterizam pelo acúmulo intralisossomal de GAGs de produtos que não foram degradados devido à falta de enzimas [1]. A MPS tipo I, também conhecida como síndrome de Hurler, se dá pela deficiência da enzima α-Liduronidase do cromossomo 4p16.3, que é responsável pela degradação dos GAGs de sulfato de dermatan e sulfato de heparan, acarretando acúmulo patológico dos mesmos no organismo.2,3 Esta é a forma mais grave da doença, resultando em morte prematura, por volta dos dez anos de idade e se caracteriza pela ocorrência de hérnias inguinais, hepatoesplenomegalia, fácies grosseira, opacidade corneana, macroglossia, baixa estatura e displasia esquelética, além de sintomas respiratórios e dificuldade de alimentação, muitas vezes com início desde o nascimento e que passam despercebidos pelos pais e médicos.1-5
Na síndrome de Hurler, pacientes exibem sinais precoces (figura 2) que, infelizmente, não são identificados como parte da doença, o que poderia resultar em diagnóstico e tratamento precoce. De fato, há uma demora entre sinais e sintomas iniciais e diagnóstico dessa doença, como o ocorrido com a paciente do relato, semelhantemente ao que ocorre em outras populações.2

Figura 2. Sintomas de início e idade na síndrome de Hurle. Fonte: Extraído e modificado de BRIDGET Kiely T. et al. Early disease progression of Hurler syndrome. Orphanet Journal of Rare Diseases 2017;12(1):32.
Alguns trabalhos sugerem que, embora a idade média do início dos sintomas identificáveis para Hurler seja em torno dos seis meses de idade, a média de idade do início do tratamento é normalmente em torno de 30 a 36 meses de idade.6 No caso da paciente do relato, observou-se que foi levada ao ortopedista aos 15 meses de idade devido à deformidade em geno varo dos membros inferiores, o qual encaminhou a mesma a um geneticista e, após cinco meses, houve diagnóstico confirmado da doença.
Devido ao não reconhecimento da síndrome pelos médicos, por ser uma doença rara, e ao fato de os primeiros sinais e sintomas serem inespecíficos e sugestivos de outras doenças, o diagnóstico correto permanece atrasado e médicos de várias outras especialidades são consultados inicialmente,1,2 antes de o paciente ser encaminhado ao médico especialista na doença.
Alterações cardiovasculares são comuns nas MPSs.1,2 Na MPS I, observam-se comumente, como manifestações cardiovasculares, cardiomiopatia, hipertensão arterial pulmonar e insuficiência cardíaca.2 Na paciente relatada, observou-se hipertrofia de ventrículo esquerdo em ecocardiograma. Além disso, a paciente apresentava infecções de vias aéreas de repetição, o que também é característico da doença. A maior causa de óbito relacionada à doença, se não tratada, se dá por insuficiência cardíaca e respiratória, além de neurodegeneração devido ao acúmulo de GAGs.1
A paciente mencionada, aos 56 meses de idade, mesmo sob TRE, necessitou realizar a traqueostomia devido a complicações de seu quadro respiratório e dificuldade de intubação durante procedimento cirúrgico. De fato, vários trabalhos sugerem que, quanto mais precoce o início do TRE, pode-se minimizar os efeitos da progressão da doença e evitar sequelas irreversíveis.7
Os tratamentos atualmente disponíveis para a MPS I incluem o TCTH e a TER.7,8 Mesmo que o TCTH tenha se tornado o padrão ouro para o tratamento da MPS I, por ser o único tratamento que tem sucesso em interromper a progressão do atraso cognitivo e também atuar em outros órgãos e sistemas no corpo retardando a progressão dos danos devidos à deposição de GAGs, ele tem suas particularidades (vide figura 3), e seu procedimento ainda possui uma alta taxa de mortalidade [9,10].
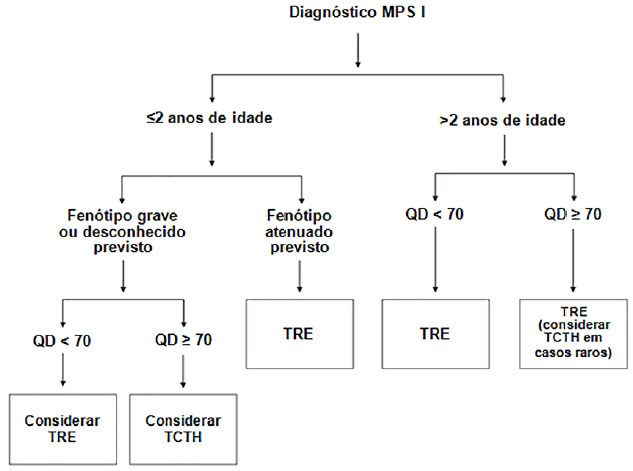
Figura 3. Algoritmo para condutas em MPS I. Fonte: Extraído e modificado de MUENZER Joseph et al. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics 2009;123(1).
A TRE consiste na aplicação periódica de uma enzima recombinante (Laronidase) por via intravenosa. Hoje em dia existem ensaios clínicos que confirmam sua eficácia e segurança no tratamento de transtornos progressivos e multissistêmicos. No caso da MPS, a maior parte das enzimas infundidas são entregues ao fígado, baço, rim e uma pequena parte pode atingir cartilagem óssea e os olhos. Porém, devido a dificuldade de as enzimas atravessarem a barreira hematoencefálica, podem ser descartados os benefícios da TRE para o desenvolvimento do sistema nervoso central.7-10
O diagnóstico precoce da doença é um fator importante, pois assim que diagnosticados, os pacientes já devem começar o tratamento com reposição enzimática para melhora da qualidade de vida e diminuição da progressão dos sintomas da doença.7-10
No momento, este tipo de tratamento constitui uma modalidade terapêutica eficaz para combater o acúmulo de GAGs e, consequentemente, controlar as implicações adversas do depósito dessa substância nos diferentes tecidos humanos.7
O tratamento com a enzima recombinante - a partir dos estudos clínicos realizados para sua aprovação e estudos posteriores de follow-up de pacientes evidenciou melhora da capacidade respiratória dos pacientes e do endurance (refletidos na melhora dos testes de caminhada e espirometria), melhora da sintomatologia da apneia obstrutiva do sono nos pacientes com maior comprometimento da capacidade respiratória, diminuição dos GAGs urinários e da visceromegalia (o que também contribui para melhoria da função cardiorrespiratória), além de melhora da função cardíaca (fração de ejeção) e da cardiomiopatia.7-11
A partir de todas as evidências, a paciente relatada iniciou a TRE aos 27 meses de idade, apresentando melhora significativa: houve aumento da estatura da paciente, diminuição da hepatoesplenomegalia e o desenvolvimento intelectual continuou o mesmo, sem alterações. Nesse caso, a reposição enzimática é feita de 7 em 7 dias, em uma dose de 15 unidades, e a única contr-indicação é que se a mesma estiver com quadro febril, não pode ser realizado o procedimento.
CONCLUSÃO
Com isso, podemos observar que o reconhecimento do conjunto de sintomas da MPS I e o encaminhamento imediato a um centro de atendimento multidisciplinar com experiência em distúrbios do armazenamento lisossomal são essenciais. Cada paciente com MPS I é único e pode apresentar um curso clínico distinto. Consequentemente, não existe uma estratégia de intervenção ou gestão de "tamanho único para todos". O diagnóstico precoce é crucial para os melhores resultados terapêuticos, sejam eles com TRE ou TCTH.
REFERÊNCIAS
1. Bruni S, Lavery C, Broomfield A. The diagnostic journey of patients with mucopolysaccharidosis I: A real-world survey of patient and physician experiences. Mol Genet Metab Rep. 2016;8:67-73.
2. Kiely BT, Kohler JL, Coletti HY, Poe MD, Escolar ML. Early disease progression of Hurler syndrome. Orphanet J Rare Dis. 2017;12(1):32.
3. Sur, Arindam et al. Mucopolysaccharidoses1(MPS1): An Atypical presentation: A Clinical Case Report. Biomedical Journal of Scientific and Technical Research 6 (2018): 001-003.
4. Stapleton M, Arunkumar N, Kubaski F, Mason RW, Tadao O, Tomatsu S. Clinical presentation and diagnosis of mucopolysaccharidoses. Mol Genet Metab. 2018;125(1-2):4-17.
5. Thakur, A.R., Naikmasur, V.G. & Sattur, A. Hurler syndrome: orofacial, dental, and skeletal findings of a case. Skeletal Radiol 44, 579-586 (2015).
6. Bruni S, Lavery C, Broomfield A. The diagnostic journey of patients with mucopolysaccharidosis I: A real-world survey of patient and physician experiences. Mol Genet Metab Rep. 2016;8:67-73.
7. Parini R, Deodato F, Di Rocco M, et al. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J Rare Dis. 2017;12(1):112.
8. 8. Muenzer J, Wraith JE, Clarke LA; International Consensus Panel on Management and Treatment of Mucopolysaccharidosis I. Mucopolysaccharidosis I: management and treatment guidelines. Pediatrics. 2009;123(1):19-29.
9. Aldenhoven M, Wynn RF, Orchard PJ, et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: an international multicenter study. Blood. 2015;125(13):2164-2172. 9.
10. Pal AR, Langereis EJ, Saif MA, et al. Sleep disordered breathing in mucopolysaccharidosis I: a multivariate analysis of patient, therapeutic and metabolic correlators modifying long term clinical outcome. Orphanet J Rare Dis. 2015;10:42.
11. Galimberti C, Madeo A, Di Rocco M, Fiumara A. Mucopolysaccharidoses: early diagnostic signs in infants and children. Ital J Pediatr. 2018 Nov 16;44(Suppl 2):133.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()