Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(4) - Dezembro 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Gustavo Lopes Nascimento
- Isabella Carramona Gonçalves
- Nicolas Henrique Cassalho Uemura
- Tatiana Oliveira Tanaca
- Palloma Bianca Ramirez Urizzi
- Laura Vagnini
- Regina Albuquerque
- Patricia Barros Viegas Anno
- Debora de Cassia Tomaz
- Marcela Almeida
- Jacqueline Fonseca
- Zumira Aparecida Carneiro
- Ana Paula Andrade Hamad
- Paula Maria Preto Mimura
- Charles Marques Lourenco
Relato de Caso
Fenótipo Atenuado da Síndrome de Hunter (Mucopolissacaridose tipo II): Desafios no Diagnóstico Clínico
Attenuated Phenotype in Hunter Syndrome (Mucopolysaccharidosis type II): Challenges in Clinical Diagnosis
Gustavo Lopes Nascimento1; Isabella Carramona Gonçalves1; Nicolas Henrique Cassalho Uemura1; Tatiana Oliveira Tanaca1; Palloma Bianca Ramirez Urizzi1; Laura Vagnini2; Regina Albuquerque3; Patricia Barros Viegas Anno3; Debora de Cassia Tomaz3; Marcela Almeida1; Jacqueline Fonseca4; Zumira Aparecida Carneiro1; Ana Paula Andrade Hamad5; Paula Maria Preto Mimura6; Charles Marques Lourenco1
DOI:10.31365/issn.2595-1769.v20i4p155-161
1. Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirao Preto - São Paulo - Brasil
2. Centro Paulista de Diagnostico e Pesquisa, Genetica Clínica - Ribeirão Preto - SP - Brasil
3. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
4. Laboratório DLE, Genética Bioquímica - Rio de Janeiro - RJ - Brasil
5. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - Ribeirão Preto - SP - Brasil
Endereço para correspondência:
Caio Bruno Andrade Nascimento
Recebido em: 29/02/2020
Aprovado em: 22/09/2020
Instituição: Universidade do Estado de Minas Gerais, Faculdade de Medicina - Passos - Minas Gerais - Brasil
Resumo
INTRODUÇÃO: Formas atenuadas de mucopolissacaridose (MPS) podem ser facilmente negligenciadas devido à escassez de sinais clínicos. Os achados cardiovasculares podem ser diagnosticados erroneamente como doença valvar devido à "febre reumática". Diferentemente da maioria dos pacientes com mucopolissacaridoses, os indivíduos com inteligência normal (fenótipo atenuado) e organomegalia leve ou ausente. Relatamos caso de paciente do sexo masculino, 7 anos de idade, com estatura normal, leve rigidez distal dos dedos, inteligência normal, doença valvar leve cujo primeiro diagnóstico foi febre reumática, embora nenhum achado laboratorial óbvio pudesse confirmar essa hipótese. A avaliação dermatológica para lesão da pele sobre as escápulas também falhou em levar a um diagnóstico. Uma avaliação clínica no serviço de genética médica apontou para suspeita de forma atenuada de MPS II, confirmada por ensaio enzimático, cromatografia de glicosaminoglicanos (GAGs) e análise molecular do gene IDS. O início relativamente tardio da doença e os sinais clínicos ausentes e /ou mínimos nas formas atenuadas apresentam grandes dificuldades para os médicos suspeitarem de tal condição. Os profissionais da área de saúde em todas as especialidades devem ter em conta que os pacientes com Hunter atenuado podem apresentar características não clássicas como doença valvular ligeira ou uma condição dermatológica. Diagnóstico e tratamento precoces melhoram significativamente o prognóstico e as atividades diárias desses pacientes.
Palavras-chave: Mucopolissacaridose II; Iduronato Sulfatase; Terapia de Reposição de Enzimas; Diagnóstico Diferencial; Glicosaminoglicanos.
Abstract
INTRODUÇÃO: Attenuated forms of MPS can be easily overlooked because of paucity of clinical signs. Moreover, they are often misdiagnosed as Legg-Calve-Perthes disease or skeletal dysplasia in presence of prominent skeletal abnormalities and lack of involvement of other systems. Cardiovascular findings can be misdiagnosed as valve disease due to "rheumatic fever". Unlike most patients with mucopolysaccharidoses (MPS), individuals with Hunter syndrome (MPS type II) do not have corneal clouding and can present with normal intelligence (attenuated phenotype) and mild/no organomegaly. Here we report a 7-year-old boy with normal stature, mild distal fingers stiffness, normal intelligence, mild valve disease whose first diagnosis was rheumatic fever, although no obvious laboratory finding could confirm this hypothesis. Dermatological evaluation for pebbling of the skin over the scapulae also failed in lead to a diagnosis. Clinical evaluation in medical genetics service pointed out to suspicion of mild Hunter syndrome, later confirmed by enzyme assay, GAGs chromatography and molecular analysis of IDS gene.In addition, relatively late onset of the disease and absent/minimal clinical signs in the attenuated forms pose great difficulties for the clinicians to suspect such condition. Healthcare providers across specialties should have in account that mild Hunter patients can present with non-classical features as mild valve disease or a dermatological condition. Early diagnosis and early treatment significantly improve the clinical outcome and activity of daily living in such patients.
Keywords: Mucopolysaccharidosis II; Iduronate Sulfatase; Enzyme Replacement Therapy; Diagnosis; Differential; Glycosaminoglycans.
INTRODUÇÃO
As mucopolissaridoses (MPS) são doenças genético-metabólicas nas quais tem-se uma alteração estrutural das células na porção dos mucopolissacarídeos, que são longas cadeias de moléculas de açúcar responsáveis pela construção dos ossos, cartilagens e tecidos do corpo1. Por definição, os mucopolissacarídeos também são chamados de glicosaminoglicanos (GAGs). Pacientes portadores de MPS acumulam GAGs e cada GAG é responsável por um subtipo da doença, entre eles o dermatan sulfato, heparan sulfato e queratan sulfato. A herança genética da síndrome de Hunter está ligada ao cromossomo X, dessa forma a maioria das mulheres são portadoras assintomáticas, em decorrência do fenômeno da inativação randômica do cromossomo X1,2. A mucopolissacaridose tipo II (MPS II) ou síndrome de Hunter é uma doença rara, uma vez que a incidência está em torno de 0,84 casos para cada 100.000 indivíduos. Entretanto, o subtipo II é o mais frequente entre todos os seis subtipos2,3,4,5.
Em geral, a apresentação clínica da síndrome de Hunter (MPS II) não está presente ao nascer, mas é comum aparecer após o primeiro ano de vida. Geralmente, os primeiros sinais e sintomas da síndrome incluem hérnias inguinais, infecções de ouvido, coriza e resfriados1,4. À medida que o acúmulo de GAGs se intensifica em todas as células do corpo, os sinais da síndrome de Hunter se tornam mais visíveis e crônicos. As manifestações físicas apresentadas por muitas crianças incluem características faciais grosseiras típicas, entre elas testa saliente, ponte nasal rebaixada (nariz em "sela"), macroglossia, macrocefalia e abdômen proeminente6,7. Todas as articulações importantes, incluindo pulsos, cotovelos, ombros, quadris e joelhos, podem ser afetadas pela síndrome de Hunter, o que leva à rigidez das articulações e limitações de mobilidade. Alguns portadores da MPS II podem apresentar, ainda, lesões cutâneas em braços, pernas e costas8,9.
O prognóstico de MPS II está diretamente relacionado ao grau de apresentação da doença, comorbidades prévias do paciente e até mesmo o acesso ao tratamento com reposição enzimática10,11,12. Atualmente, não é possível determinar, em anos, a sobrevida de um paciente portador da síndrome, mas pode-se afirmar que quanto mais severa for a forma da doença, pior o prognóstico e consequentemente menor será a expectativa de vida8,11.
O objetivo do trabalho é alertar os profissionais da saúde sobre as formas atenuadas da doença que podem passar despercebidas, retardando o diagnóstico e, consequentemente, o início do tratamento, fator que interfere diretamente no prognóstico e na qualidade de vida desses pacientes. Neste relato, os dados foram obtidos através da revisão do prontuário com aautorização do comitê de ética do hospital e do paciente, obtido por assinatura do termo de consentimento livre e esclarecido (CAAE 70623317.7.0000.5581)
2. RELATO DE CASO
Paciente sete anos, filho de casal jovem e não-consanguíneo. Aos 4 anos foi levado ao pediatra com queixa de cansaço e dispneia, dando sequência a maiores investigações devido suspeita clínica de valvulopatia cardíaca. Após consulta com especialista, o paciente foi diagnosticado com febre reumática e o tratamento recomendado consistia em seguimento até os 18 anos com injeções de Penicilina benzatina a cada 21 dias. Embora paciente não tivesse critérios laboratoriais (reagentes de fase aguda como velocidade de hemossedimentação e proteína C reativa eram normais) ou mesmo clínicos (ausência de artrite, coreia, nódulos subcutâneos, aumento de intervalo PR, eritema marginatum ou de episódios de febre prévios) para esse diagnóstico.
Histórico gestacional sem intercorrências, no entanto a genitora não fez exames pré- natais. Relato de boa movimentação fetal (a partir do 3o mês de gestação). Nascido de parto cesáreo a termo, pesando 3.300g e com comprimento de 49 cm. Obteve alta com dois dias de vida. Apresentou nos primeiros anos de vida, infecções de repetição das vias aéreas superiores, incluindo otites de repetição. Diversos episódios de infecções de ouvido e de vias aéreas superiores que foram clinicamente tratados.
Desenvolvimento neuropsicomotor normal. Sentou com sete meses, andou com um ano, mesma época falou as primeiras palavras. Com cerca de cinco anos, a família observou que a criança apresentava rigidez articular em mãos e pescoço, sendo encaminhada para avaliação com pediatra e fisioterapeuta, sem elucidação diagnóstica. Nega história familiar de doenças reumáticas, ou doenças autoimunes.
Por volta dos cinco anos de idade, surgiram lesões cutâneas periescapulares bilaterais (Figura 1), de etiologia desconhecida. Aos 6 anos, foi avaliado por um ortopedista por apresentar marcha na ponta dos pés (com posterior resolução espontânea). Paciente foi submetido a um procedimento cirúrgico para correção de uma hérnia presente desde o nascimento. A mãe do paciente relata que todas as alterações que surgiam eram tratadas com especialistas para correção do problema pontual, sem que houvesse um diagnóstico conclusivo a respeito da causa desses sintomas, aparentemente não relacionados.
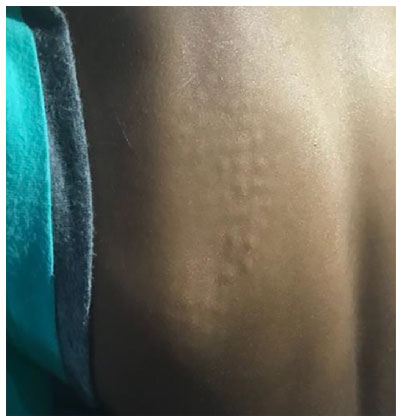
Figura 1. Lesões periescapulares por infiltrados de GAGs
A hipótese diagnóstica surgiu de maneira informal quando familiares visualizaram um cartaz informativo, no qual o menino da propaganda, portador de uma doença de armazenamento lisossômico, era fisicamente muito parecido com o paciente e apresentava as mesmas alterações recorrentes, levantando, assim, à suspeita dessa enfermidade. Em vista disso, paciente foi encaminhado para avaliação com médico geneticista.
Seu exame genético-clínico revelou estatura no percentil 3, peso entre os percentis 25 e 50, perímetro cefálico entre os percentis 50 e 97, face discretamente infiltrada, dorso nasal curto, raiz nasal achatada (Figura 2), contratura e rigidez das articulações, mãos em garra (Figura 3), presença de hérnia umbilical, reflexos osteotendíneos grau 2 em membro inferior e superior e ausência de sinais de compressão medular.

Figura 2. Aumento do infiltrado de GAGs evolutivamente: (A) 1 ano, (B) 3 anos C) 6 anos de idade.
Fonte.Os autores (2018)
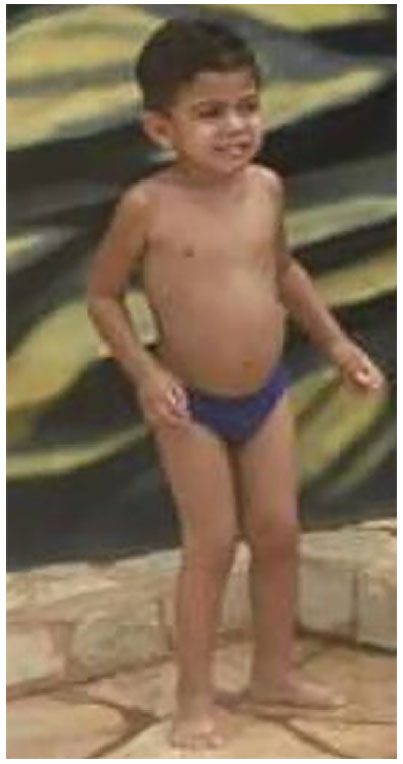
Figura 3. Contratura e rigidez articular em membros superiores
Fonte.Os autores (2018)
Devido à presença de achados sugestivos de mucopolissacaridose, o geneticista orientou a realização de exames específicos para confirmação da hipótese diagnóstica. Foram solicitados exames complementares para melhor elucidação do caso: exames enzimáticos, radiografia de coluna tóraco-lombar, de mãos e de pés e cromatografia/dosagem de glicosaminoglicanos na urina (Tabelas 1 e 2).
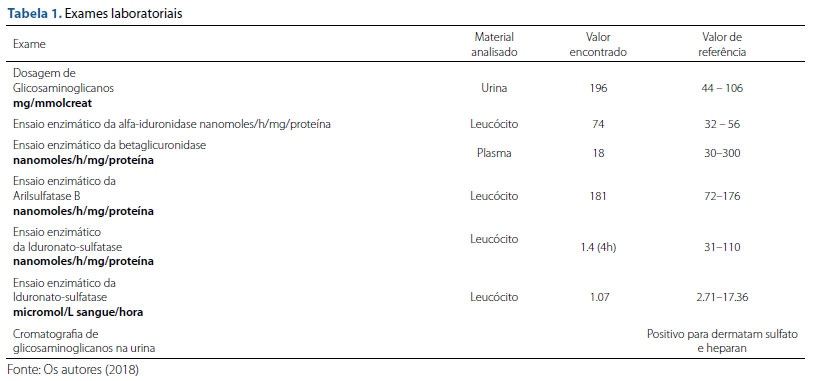

A tabela 1 mostra a presença de níveis significativamente aumentados para concentração de glicosaminoglicanos com presença de dermatan e heparan sulfato em cromatografia de glicosaminoglicanos, contudo, para o diagnóstico definitivo, é essencial a confirmação da da hipótese diagnóstica a realização dos testes enzimáticos específicos que evidenciaram deficiência da iduronato-sulfatase.
Além disso, o exame clínico do paciente, associado aos exames complementares, foram compatíveis com o diagnóstico de Mucopolissacaridose tipo II, também conhecida como síndrome de Hunter. Teste genético molecular feito posteriormente com sequenciamento do gene IDS evidenciando a presença da mutação missense (p.Asn63SAsp) no exon 2, confirmando que o paciente é hemizigoto para dada mutação, a qual também foi identificada em sua genitora assintomática.
DISCUSSÃO
Os glicosaminoglicanos (GAGs) são longas cadeias de moléculas de açúcar usadas na construção de ossos, cartilagem, pele, tendões e outros tecidos do corpo. Com o acúmulo progressivo dos GAGs, pode advir comprometimento de diversos órgãos, particularmente o encéfalo13,14. Em uma célula saudável os GAGs se soltam da membrana e entram dentro dos lisossomos para serem "reciclados" na qual tem-se o envolvimento de uma série de enzimas. A cadeia das GAGs é desfeita a partir da remoção de uma molécula de açúcar por vez em uma das pontas da cadeia15,16. Indivíduos portadores de MPS II apresentam uma deficiência ou anormalidade na enzima iduronato-2-sulfatase (I2S)17 responsável pela quebra das GAGs (dermatan sulfato e heparan sulfato), como o material não é degradado ocorre um acúmulo dos mesmos no interior dos lisossomos causando um aumento significativo de tamanho e consequentemente do funcionamento da célula18,19.
À medida que o acúmulo de GAGs se intensifica em todas as células do corpo, os sinais da síndrome de Hunter se tornam mais visíveis e crônicos20,21. As manifestações físicas apresentadas por muitas crianças incluem características faciais grosseiras típicas, entre elas testa saliente, ponte nasal rebaixada (nariz em "sela"), macroglossia, macrocefalia e abdômen proeminente. Por isso, é comum que as crianças com a síndrome de Hunter sejam parecidas entre si, mesmo quando não tenham grau de parentesco22.
A despeito disso, achados faciais podem ser sutis em alguns pacientes com síndrome de Hunter, contribuindo para o não reconhecimento dessa enfermidade, já que muitos clínicos e pediatras associam pacientes com mucopolissacaridoses ao facies grosseiro23. O paciente em questão, embora seja possível observar leves características faciais que poderiam sugerir a enfermidade, pode ser considerado como portador de uma forma atenuada da doença, especialmente porque o mesmo apresenta inteligência compatível com a normalidade, o que pode ocorrer em até 40% dos pacientes com síndrome de Hunter23.
No decorrer dos anos, as otites e amigdalites tornam-se mais frequentes, tem-se o surgimento de valvulopatias devido ao espessamento das válvulas e das paredes cardíacas, problemas respiratórios com obstrução de vias aéreas e capacidade pulmonar limitada23,24. O tipo de valvulopatia presente nos pacientes com síndrome de Hunter pode mimetizar o padrão observado na valvulopatia reumática, já que, em ambas as enfermidades, ocorre alteração do tecido conectivo valvular, tornando-se um fator de confusão no diagnóstico de mucopolissacaridose em pacientes que não apresentam fenótipo facial típico, como observado no paciente do relato.
Com a progressão da doença, é possível identificar uma hepatoesplenomegalia, tal condição faz com que as hérnias fiquem mais evidentes devido à distensão da parede abdominal25, muito embora as hérnias possam estar presentes mesmo na ausência de visceromegalia
As grandes articulações, incluindo pulsos, cotovelos, ombros, quadris e joelhos podem ser afetadas pela síndrome de Hunter, o que leva à rigidez das mesmas e limitações de mobilidade26. A marcha na ponta dos pés pode representar encurtamento dos tendões aquileus e é também parte do espectro osteoarticular da mucopolissacaridose tipo II26.
Alguns portadores da MPS II podem apresentar lesões cutâneas em braços, pernas e costas27, podendo ser uma das pistas mais específicas para síndrome de Hunter, já que não estão presentes em outros tipos de mucopolissacaridose.
O armazenamento de GAGs no cérebro pode levar o paciente a um atraso de desenvolvimento com subsequente deficiência intelectual28,29. Quando o predomínio do acúmulo é ocular, o paciente passa comumente apresentar retinopatias, como papiledema30,31,32. A velocidade e o grau de progressão da doença podem diferir de pessoa para pessoa, o que apresenta ampla variabilidade quanto à gravidade de sintomas. É importante observar que embora o termo "brando/atenuado" seja usado pelos médicos ao comparar os pacientes com a síndrome de Hunter, os efeitos da doença são sérios e limitantes. Essa variabilidade de espectro de gravidade da doença determinará a presença e o grau de deficiência mental e a expectativa de vida do paciente10.
A MPS II costumava ser dividida em dois grandes grupos (grave e leve). Atualmente, ela pode ser classificada em MPS II-grave, MPS II-intermediária e MPS II-leve, de acordo com a magnitude dos sintomas apresentados pelos pacientes. Os indivíduos com MPS II-grave têm atraso de desenvolvimento progressivo e problemas físicos mais graves. Os indivíduos com MPS II-leve/atenuada, que é o caso do nosso paciente, têm inteligência normal, problemas físicos mais leves e sobrevida praticamente normal. Na forma intermediária, os indivíduos apresentam inteligência normal ou pouco comprometida) e problemas físicos mais severos. Todas as pessoas com MPS II têm a mesma enzima faltando, independente da gravidade do quadro clínico associado. A forma grave é relatada como sendo três vezes mais frequente que a leve33.
Os indivíduos com MPS II frequentemente têm acometimentos sistêmicos, tais como infecções crônicas de ouvido, problemas de visão e de audição, rigidez de articulações23, cardiopatia (tanto cardiomiopatia quanto valvulopatia envolvendo as válvulas aórtica e mitral), hérnias inguinal e umbilical24, síndrome do túnel do carpo25, hidrocefalia comunicante e apneia do sono 26,27,28 necessitando de acompanhamento multidisciplinar.
A associação de uma dieta específica para a redução do acúmulo de GAGs não está intimamente ligada à melhora da MPS II. Alguns pacientes relatam melhora em pontos específicos como a diarreia, quando se evita a ingestão de leite, porém o mesmo não se aplica a todos os sinais e sintomas da doença34. Com o avançar da doença, o uso de sondas nasogástricas ou até mesmo gastrostomias tornam-se necessárias devido ao prejuízo da deglutição e consequentemente broncoaspiração e posterior quadro de pneumonia aspirativa35.Para o tratamento dos quadros de rigidez articular, são indicadas sessões de fisioterapia com significativas melhoras em casos leves ou moderados de limitação. Pacientes com limitações mais graves, entretanto, não se beneficiarão das sessões de fisioterapia no sentido de amenizar as restrições articulares, mas sim como forma preventiva de evolução da rigidez articular36.
O objetivo do tratamento da MPS II é reduzir a progressão da doença, evitar acometimentos de órgãos nobres e proporcionar uma boa qualidade de vida aos pacientes. Até alguns anos atrás, tratava-se apenas os sintomas da doença, mas, desde 2006, vem-se fazendo um tratamento específico baseado em uma terapia de reposição enzimática (TRE), na qual são feitas infusões intravenosas semanais da enzima idursulfase (Elaprase®). Essa terapia auxilia na diminuição dos acúmulos de GAGs no interior dos lisossomos, e consequentemente, diminuir o acúmulo em órgãos como fígado, baço, vias respiratórias superiores, músculo cardíaco, melhora o padrão de sono e a mobilidade articular37,38.
Assim, é possível concluir que o paciente em questão possui a forma leve/atenuada da síndrome de Hunter, não apresentando qualquer problema no desenvolvimento neurológico e de aprendizagem. Suas manifestações clínicas foram mais restritas aos sintomas osteoarticulares, cardiovasculares e cutâneos39. No entanto, dado o caráter progressivo da enfermidade, espera-se que, na ausência de terapia específica, o paciente possa vir apresentar outras complicações associadas à doença, como piora da rigidez articular, sintomas associados ao túnel do carpo bilateral, progressão da valvulopatia, perda auditiva, entre outros sintomas40. Dessa forma, não há dúvidas de que o início precoce do tratamento é de suma importância para o desenvolvimento e melhor qualidade de vida desses pacientes.
Mesmo na forma leve/atenuada da doença, deve-se sempre levar em conta que, a longo prazo, o acúmulo das GAGs acarreta limitações significativas na vida dos pacientes, como também piora da sua qualidade de vida e de seus cuidadores41. Por conseguinte, o diagnóstico precoce é fundamental e, para que isso seja possível, clínicos, pediatras, ortopedistas, cardiologistas, cirurgiões pediátricos, neuropediatras e otorrinolaringologistas devem estar atentos às manifestações incipientes dessa enfermidade, como as apresentadas por nosso paciente, visto que um diagnóstico precoce pode permitir iniciar terapia específica antes que sequelas irreversíveis da enfermidade estejam presentes.
REFERÊNCIAS
1. Giugliani R. Mucopolysacccharidoses: From understanding to treatment, a century of discoveries. Genet Mol Biol. 2012 Dec. 35 (4 (suppl)):924-31.
2. Timms KM, Lu F, Shen Y, et al. 130 kb of DNA sequence reveals two new genes and a regional duplication distal to the human iduronate-2-sulfate sulfatase locus. Genome Res. 1995 Aug. 5(1):71-8..
3. Lowry RB, Applegarth DA, Toone JR, MacDonald E, Thunem NY. An update on the frequency of mucopolysaccharide syndromes in British Columbia. Hum Genet. 1990 Aug. 85(3):389-90.
4. Timms KM, Bondeson ML, Ansari-Lari MA, et al. Molecular and phenotypic variation in patients with severe Hunter syndrome. Hum Mol Genet. 1997 Mar. 6(3):479-86. .
5. Young ID, Harper PS. Incidence of Hunter's syndrome. Hum Genet. 1982. 60(4):391-2.
6. Matern D. Newborn screening for lysosomal storage disorders. Acta Paediatr Suppl. 2008 Apr. 97(457):33-7.
7. Schaap T, Bach G. Incidence of mucopolysaccharidoses in Israel: is Hunter disease a "Jewish disease"?.Hum Genet. 1980. 56(2):221-3.
8. Young ID, Harper PS. The natural history of the severe form of Hunter's syndrome: a study based on 52 cases. Dev Med Child Neurol. 1983 Aug. 25(4):481-9.
9. Hobolth N, Pedersen C. Six cases of a mild form of Hunter syndrome in five generations. Three affected males with progeny. Clin Genet. 1978. 20:121.
10. Young ID, Harper PS, Newcombe RG, Archer IM. A clinical and genetic study of Hunter's syndrome. 2. Differences between the mild and severe forms. J Med Genet. 1982 Dec. 19(6):408-11.
11. Young ID, Harper PS. Mild form of Hunter's syndrome: clinical delineation based on 31 cases. Arch Dis Child. 1982 Nov. 57(11):828-36.
12. Schulze-Frenking G, Jones SA, Roberts J, Beck M, Wraith JE. Effects of enzyme replacement therapy on growth in patients with mucopolysaccharidosis type II. J Inherit Metab Dis. 2011 Feb. 34(1):203-8.
13. Brusius-Facchin AC, Abrahão L, Schwartz IV, Lourenço CM, Santos ES, Zanetti A, et al. Extension of the molecular analysis to the promoter region of the iduronate 2-sulfatase gene reveals genomic alterations in mucopolysaccharidosis type II patients with normal coding sequence. Gene. 2013 Sep 10. 526(2):150-4.
14. Isogai K, Sukegawa K, Tomatsu S, et al. Mutation analysis in the iduronate-2-sulphatase gene in 43 Japanese patients with mucopolysaccharidosis type II (Hunter disease). J Inherit Metab Dis. 1998 Feb. 21(1):60-70.
15. Garcia AR, Pan J, Lamsa JC, Muenzer J. The characterization of a murine model of mucopolysaccharidosis II (Hunter syndrome). J Inherit Metab Dis. 2007 Sep 16.
16. Friso A, Tomanin R, Alba S, Gasparotto N, Puicher EP, Fusco M, et al. Reduction of GAG storage in MPS II mouse model following implantation of encapsulated recombinant myoblasts. J Gene Med. 2005 Nov. 7 (11):1482-91.
17. Crotty PL, Braun SE, Anderson RA, Whitley CB. Mutation R468W of the iduronate-2-sulfatase gene in mild Hunter syndrome (mucopolysaccharidosis type II) confirmed by in vitro mutagenesis and expression.HumMol Genet. 1992 Dec. 1(9):755-7.
18. Birot AM, Delobel B, Gronnier P, Bonnet V, Maire I, Bozon D. A 5-megabase familial deletion removes the IDS and FMR-1 genes in a male Hunter patient. Hum Mutat. 1996. 7(3):266-8.
19. Hopwood JJ, Bunge S, Morris CP, et al. Molecular basis of mucopolysaccharidosis type II: mutations in the iduronate-2-sulphatase
20. Rathmann M, Bunge S, Beck M, Kresse H, Tylki-Szymanska A, Gal A. Mucopolysaccharidosis type II (Hunter syndrome): mutation "hot spots" in the iduronate-2-sulfatase gene. Am J Hum Genet. 1996 Dec. 59(6):1202-9.
21. Clarke JT, Wilson PJ, Morris CP, et al. Characterization of a deletion at Xq27-q28 associated with unbalanced inactivation of the nonmutant X chromosome. Am J Hum Genet. 1992 Aug. 51(2):316-22.
22. Spranger J, Cantz M, Gehler J, Liebaers I, Theiss W. Mucopolysaccharidosis II (Hunter disease) with corneal opacities. Report on two patients at the extremes of a wide clinical spectrum. Eur J Pediatr. 1978 Aug 17. 129(1):11-6.
23. Rozdzynska A, Tylki-Szymanska A, Jurecka A, Cieslik J. Growth pattern and growth prediction of body height in children with mucopolysaccharidosis type II. ActaPaediatr. 2011 Mar. 100(3):456-60.
24. Kamin W. Diagnosis and management of respiratory involvement in Hunter syndrome. ActaPaediatr Suppl. 2008 Apr. 97(457):57-60.
25. Kampmann C, Beck M, Morin I, Loehr JP. Prevalence and characterization of cardiac involvement in hunter syndrome. J Pediatr. 2011 Aug. 159(2):327-331.e2.
26. Mullen CA, Thompson JN, Richard LA, Chan KW. Unrelated umbilical cord blood transplantation in infancy for mucopolysaccharidosis type IIB (Hunter syndrome) complicated by autoimmune hemolytic anemia.Bone Marrow Transplant. 2000 May. 25(10):1093-7.
27. Kwon JY, Ko K, Sohn YB, et al. High prevalence of carpal tunnel syndrome in children with mucopolysaccharidosis type II (Hunter syndrome). Am J Med Genet A. 2011 Jun. 155(6):1329-35.
28. Neufeld EF, Muenzer J. The Mucopolysaccharidoses. The Metabolic Bases of Inherited Disease. 8th ed. McGraw-Hill; 2000. 3421-52.
29. Holt J, Poe MD, Escolar ML. Early Clinical Markers of Central Nervous System Involvement in Mucopolysaccharidosis Type II. J Pediatr. 2011 Aug. 159(2):320-326.e2.
30. Wraith JE, Beck M, Giugliani R, Clarke J, Martin R, Muenzer J. Initial report from the Hunter Outcome Survey. Genet Med. 2008 Jul. 10(7):508-16.
31. Caruso RC, Kaiser-Kupfer MI, Muenzer J, Ludwig IH, Zasloff MA, Mercer PA. Electroretinographic findings in the mucopolysaccharidoses. Ophthalmology. 1986 Dec. 93(12):1612- 6.
32. Beck M. Papilloedema in association with Hunter's syndrome. Br J Ophthalmol. 1983 Mar. 67(3):174-7.
33. Beck M, Cole G. Disc oedema in association with Hunter's syndrome: ocular histopathological findings. Br J Ophthalmol. 1984 Aug. 68(8):590-4.
34. Clarke LA. Idursulfase for the treatment of mucopolysaccharidosis II. Expert OpinPharmacother. 2008 Feb. 9(2):311-7.
35. Chmielarz I, Gabig-Ciminska M, Malinowska M, Banecka-Majkutewicz Z, Wegrzyn A, Jakobkiewicz-Banecka J. Comparison of siRNA-mediated silencing of glycosaminoglycan synthesis genes and enzyme replacement therapy for mucopolysaccharidosis in cell culture studies. ActaBiochim Pol. 2012. 59(4):697-702.
36. Meikle PJ, Grasby DJ, Dean CJ, et al. Newborn screening for lysosomal storage disorders. Mol Genet Metab. 2006 Aug. 88(4):307-14.
37. Vellodi A, Young E, Cooper A, Lidchi V, Winchester B, Wraith JE. Long-term follow-up following bone marrow transplantation for Hunter disease. J Inherit Metab Dis. 1999 Jun. 22(5):638-48.
38. Martin R, Beck M, Eng C, et al. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome).Pediatrics. 2008 Feb. 121(2):e377-86.
39. Muenzer J, Gucsavas-Calikoglu M, McCandless SE, Schuetz TJ, Kimura A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol Genet Metab. 2007 Mar. 90(3):329-37.
40. Muenzer J, Wraith JE, Beck M, et al. A phase II/III clinical study of enzyme replacement therapy with idursulfase in mucopolysaccharidosis II (Hunter syndrome). Genet Med. 2006 Aug. 8(8):465-73.
41. [Guideline] Wraith JE, Scarpa M, Beck M, et al. Mucopolysaccharidosis type II (Hunter syndrome): a clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur J Pediatr. 2008 Mar. 167(3):267-77.
42. Al Sawaf S, Mayatepek E, Hoffmann B. Neurological findings in Hunter disease: pathology and possible therapeutic effects reviewed. J Inherit Metab Dis. 2008 Aug. 31(4):473-80.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()