Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(4) - Dezembro 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores- Vinicius Munute Desiderio
- Fernando Oliveira
- Fernando Moraes Teruel
- Isabela Guimarães
- Jacqueline Fonseca
- Laura Vagnini
- Marcela Almeida
- Regina Albuquerque
- Debora Tomaz
- Patricia Barros Viegas Anno
- Ana Paula Andrade Hamad
- Zumira Aparecida Carneiro
- Juliana Maria Faccioli Sicchieri
- Charles Marques Lourenco
Relato de Caso
Intolerância hereditária à frutose: uma causa não tão rara de hepatopatia na infância - relato de caso
Hereditary fructose intolerance: a not so rare cause of childhood liver disease - case report
Vinicius Munute Desiderio1; Fernando Oliveira1; Fernando Moraes Teruel1; Isabela Guimarães2; Jacqueline Fonseca3; Laura Vagnini4; Marcela Almeida1; Regina Albuquerque2; Debora Tomaz2; Patricia Barros Viegas Anno2; Ana Paula Andrade Hamad5; Zumira Aparecida Carneiro1; Juliana Maria Faccioli Sicchieri6; Charles Marques Lourenco1,2,4
DOI:10.31365/issn.2595-1769.v20i4p140-144
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
3. Laboratório DLE, Genética Bioquímica - Rio de Janeiro - RJ - Brasil
4. Centro Paulista de Diagnóstico e Pesquisa, Genática Clínica - Ribeirão Preto - SP - Brasil
5. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Neurociências e Ciências do Comportamento - Ribeirão Preto - São Paulo - Brasil
6. Hospital das Clínicas de Ribeirão Preto - Faculdade de Medicina de Ribeirão Preto (FMRP) - Universidade de São Paulo, Departamento de Medicina Interna - Ribeirão Preto - SP - Brasil
Endereço para correspondência:
Charles Marques Lourenco
Recebido em: 11/11/2019
Aprovado em: 06/09/2020
Instituição: Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
Resumo
INTRODUÇÃO: A intolerância hereditária à frutose (IHF) é uma rara doença caracterizada pela deficiência da enzima aldolase B codificada pelo gene ALDOB. Manifesta-se geralmente com hipoglicemia após a ingestão de alimentos ou compostos que possuem frutose em sua composição. Diagnóstico precoce e dieta rigorosa com restrição de frutose são essenciais para se evitar complicações clínicas e sequelas irreversíveis ao paciente.
CASO CLÍNICO: Apresenta-se um caso de um paciente de 6 meses que deu entrada em hospital com hipoglicemia após a ingestão de suco de fruta pela primeira vez. Foram realizados exames de ultrassom abdominal e ressonância magnética, que evidenciaram esteatose hepática. O quadro clínico foi altamente sugestivo de IHF, portanto o gene ALDOB foi solicitado para o paciente. A análise gênica mostrou a presença de duas mutações heterozigotas, herdadas de cada progenitor, confirmando o diagnóstico clínico de IHF.
CONCLUSÃO: O relato de caso pretende reforçar a importância clínica e laboratorial no diagnóstico de IHF, tal como o desfecho clínico e a importância da dietoterapia em seu tratamento.
Palavras-chave: Intolerância à frutose. Hepatopatia gordurosa não alcoólica. Erros inatos do metabolismo dos carboidratos. Erros inatos do metabolismo da frutose.
Abstract
INTRODUÇÃO: Hereditary fructose intolerance (HFI) is a rare disease characterized by deficiency of aldolase B enzyme due to mutations in ALDOB gene. It usually manifests as hypoglycemia after ingestion of foods or compounds containing fructose. Early diagnosis and fructose-restricted diet are essential to avoid clinical complications and irreversible sequelae.
CASE REPORT: Here we report a 6-month-old patient who was admitted to a hospital with hypoglycemia after ingestion for the first time of a juice fruit. Abdominal ultrasound and magnetic resonance imaging were performed and showed liver steatosis. Clinical picture was highly suggestive of HFI, so ALDOB gene was requested for the patient. Gene analysis showed presence of two heterozygous mutations, inherited from each parent thus confirming the clinical diagnosis of HFI.
CONCLUSION: The case report intends to reinforce the clinical and laboratory importance in the diagnosis of HFI as well as the clinical outcome and the importance of diet therapy in its treatment.
Keywords: Fructose intolerance. Non-alcoholic fatty liver disease. Carbohydrate metabolism, inborn errors. Fructose metabolism, inborn errors.
INTRODUÇÃO
Umas das fontes básicas de energia consumidas pelos seres humanos se tornou a grande vilã em boa parte das pessoas ao redor do mundo,1inborn errors of carbohydrate metabolism are rare conditions. Three inborn errors are known in the pathway of fructose metabolism; (1 devido ao crescente consumo de alimentos industrializados, associados ao aumento do número de estudos relacionados à frutose e os efeitos de sua ingesta excessiva no longo prazo.2
A intolerância hereditária à frutose (IHF; OMIM 229600) é uma doença metabólica rara, autossômica recessiva, resultante da deficiência de aldolase B, que acomete cerca de 1:20.000 indivíduos, independentemente do sexo.¹ Estima-se, também, que uma em cada 50 pessoas seja portadora de um alelo mutado desse gene, por isso é provável que a IHF seja ainda mais comum em muitos indivíduos adultos que vivem subdiagnosticados. O recente conhecimento de sua base molecular permitiu identificar mutações no gene da aldolase B (localizado em 9q31.1) como causadora dessa enfermidade. Esse gene é constituído por nove éxons, que são traduzidos em uma enzima de 364 aminoácidos; já foram descritas 65 mutações no gene ALDOB.3 A IHF consiste em um erro inato do metabolismo, pois a aldolase B é responsável por clivar a frutose-1-P em duas trioses (gliceraldeído e diidroxiacetona fosfato), conforme a figura 1
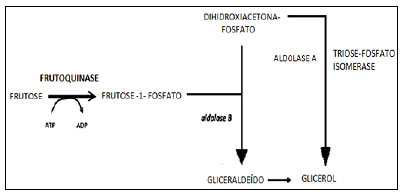
Figura 1. Conversão da Frutose pela enzima aldolase B
Gliceraldeído e diidroxiacetona fosfato são importantes em vias metabólicas de produção de energia e na lipogênese; portanto, quando o indivíduo ingere a frutose, a produção de energia através dessa via, juntamente com a lipólise, é inibida.¹ ²
A IHF pode causar lesões hepáticas, no intestino delgado e no túbulo renal proximal, podendo causar hipoglicemia, hipoalbuminemia, aumento das transaminases e bilirrubina, alteração dos fatores de coagulação, aumento dos aminoácidos séricos (metionina e tirosina), glicosúria, hiperaminoacidúria, proteinúria, acidose metabólica.4 Manifesta-se, clinicamente, com vômitos, sudorese, náuseas, dores abdominais associadas com hipoglicemia e acidose metabólica.4 Em adultos, a doença pode causar aversão "psicológica" a alimentos ricos em frutose, que ocorre de forma inconsciente após recorrentes apresentações sintomáticas - ou seja, desconforto abdominal, astenia, náuseas -, sendo que o desconforto gerado pela ingesta da frutose leva à redução da procura por alimentos que contenham esse monossacarídeo.²
Entretanto, com o abuso dos açúcares, a reação aguda é mais grave, podendo a criança ter um quadro letárgico e desenvolver convulsão ou coma.² Devido ao leite materno não conter esses açúcares (sendo composto apenas por galactose e glicose), os recém-nascidos, usualmente, não desenvolvem sintomas.5pyloric stenosis or hiatus hernia (5X O conhecimento prévio e a suspeita da IHF são a chave para o diagnóstico precoce da doença. A presença de frutose (sacarose ou sorbitol) é comum na maioria das fórmulas que são usadas em medicações infantis e vendidas sem receita médica. Além disso, diversas marcas utilizam de forma publicitária palavras como "açúcar" para designar apenas a substância sacarose em seus rótulos. Em vista disso, a falta de conhecimento dos portadores e/ou negligência ao ler os rótulos dos prudutos contendo esse composto podem levar ao atraso do diagnóstico.7 Uma boa anamnese direcionada ao histórico alimentar e aos fatores desencadeantes é essencial para esclarecer o quadro dessa síndrome.5pyloric stenosis or hiatus hernia (5X)
Caso sejam observados sinais e sintomas de IHF após o consumo de produtos contendo frutose, este deve ser suspendido imediatamente, para que o organismo tente reverter os primeiros achados de intoxicação aguda.6 A eliminação rápida e absoluta da fonte de frutose traz, consequentemente, benefício clínico ao paciente, e deve ser interpretada de forma a reforçar a suspeita diagnóstica.7 Dada a relativa inespecificidade dos atuais métodos laboratoriais, como a biópsia tecidual ou prova de sobrecarga com frutose, espera-se que, em breve, o diagnóstico genético a partir do sequenciamento do gene ALDOB se torne mais frequente, possibilitando diagnóstico mais precoce e acurado dessa enfermidade.³
Relatamos aqui o caso de um paciente com IHF cujo diagnóstico revelou-se um desafio clínico, reforçando a necessidade de os pediatras atentarem a essa possibilidade diagnóstica em pacientes com hepatomegalia e hipoglicemia após a ingestão de glicose.
RELATO DE CASO
Paciente, três anos, sexo masculino, filho de pais não consanguíneos, foi encaminhado para avaliação de quadro de hepatopatia associado a episódios de hipoglicemia.
Trata-se de paciente produto de uma gestação a termo, sem intercorrências, nascido de parto cesáreo. Ao nascimento, apresentou peso 2,795kg, comprimento de 51cm e perímetro cefálico de 35cm; APGAR 10/10. Evoluiu com leve icterícia, mas sem outros agravos à saúde, recebendo alta no segundo dia de vida. Genitores negaram quaisquer problemas de saúde no período pré-natal e nos primeiros meses de vida. Aos seis meses, contudo, apresentou grave quadro de hipoglicemia, necessitando de internação hospitalar, após a introdução do suco de fruta pela primeira vez. Genitora relatou que, após a ingesta do suco de fruta, paciente desenvolveu quadro de sonolência, culminando em hipoglicemia grave (30mg/dl) e rebaixamento do nível de consciência. Em unidade de pronto-atendimento, administrou-se glicose endovenosa e paciente foi hospitalizado para posterior investigação.
Antecedentes de marcos motores nesse período revelaram relativo atraso de desenvolvimento motor, deambulando com um ano e seis meses e leve hipotonia axial. A etiologia do quadro hipoglicêmico não foi elucidada, e em vista disso o paciente foi orientado a prosseguir investigação após alta hospitalar. Posteriormente, foi encaminhado para avaliação do endocrinologista, o qual, diante do histórico alimentar do paciente e a aparente relação temporal do quadro de hipoglicemia após a início da alimentação com fruta, levantou a hipótese diagnóstica de intolerância hereditária à frutose (IHF) e o encaminhou para serviço terciário de genética.
Ao ser avaliado nesse centro, solicitaram-se exames complementares como ultrassonografia e ressonância abdominal, as quais evidenciaram a presença de esteatose hepática. Realizou-se investigação exaustiva para outras doenças metabólicas que cursassem com hepatopatia na infância, cujos resultados revelaram-se dentro da normalidade.
Diante da não elucidação diagnóstica e da suspeita de intolerância hereditária à frutose pela história clínica, procedeu-se ao sequenciamento direto do gene ALDOB, o qual, contudo, apenas evidenciou presença de uma mutação missense patogênica: c.524C>A; p. (Ala175Asp). Para prosseguir a investigação sem necessidade de exames mais invasivos (como a biópsia de fígado), optou-se por realizar um painel de genes relacionados a hepatopatia crônica/colestase de causa genética: duas mutações, em heterozigose composta, foram identificadas no gene ALDOB, a mutação c.448G>C (p.A150P), de origem paterna, e a mutação c.524C>A(p.A175D), de origem materna, confirmando-se o diagnóstico de IHF.
Diante da elucidação etiológica do seu quadro clínico, paciente foi encaminhado para nutricionista especialista em doenças metabólicas, com a finalidade de realizar dieta restrita em frutose. Curiosamente, a essa época os genitores já observavam que o paciente apresentava rejeição a alguns alimentos sólidos contendo frutose, além de episódios de distensão abdominal após realizar, inadvertidamente, refeições contendo frutas. Em virtude disso, paciente passou a utilizar fórmulas nutricionais com restrição de frutose até o manejo dietético específico a ser realizado pelo nutricionista. Elaborou-se cardápio com presença de alimentos sólidos que o paciente segue até hoje, consistindo a dieta em oferta de alimentos de origem animal à vontade (com exceção de leite integral), além de uso de quantidades restritas de batata, feijão e lentilha.
Atualmente, aos três anos e seis meses, ao exame físico o paciente apresenta-se lúcido, orientado em tempo e espaço, com face simétrica, sem dismorfias faciais; ausência de cáries com normoimplantação dentária; movimentos oculares dentro da normalidade; força muscular preservada em todos os membros, reflexos osteotendíneos normoativos, leve hipotonia axial de predomínio de membros inferiores; leve frouxidão ligamentar, sem alteração de equilíbrio e de coordenação motora grossa ou fina, reflexo cutâneo plantar em flexão, sensibilidade preservada, marcha atípica.
DISCUSSÃO
A intolerância hereditária à frutose, doença metabólica rara descrita pela primeira vez em 1956, é caracterizada pelo erro da clivagem da frutose-1-P em duas trioses, devido à deficiência da enzima hepática aldolase B.8 Essa enzima é importante em vias metabólicas de produção de energia e na lipogênese; portanto, quando o indivíduo ingere esse tipo de açúcar, as produções de energia através dessa via, juntamente com a lipólise, são inibidas.1,3
A IHF pode cursar com lesões hepáticas, renais e intestinais, podendo apresentar-se clinicamente com náuseas, vômitos, desconforto abdominal, astenia, convulsão e coma, devido aos quadros de hipoglicemia, hipoalbuminemia, aumento de aumento das transaminases e bilirrubina, alteração dos fatores de coagulação, aumento dos aminoácidos séricos (metionina e tirosina), glicosúria, hiperaminoacidúria, proteinúria e acidose metabólica.5 O diagnóstico é dado a partir de uma anamnese direcionada ao histórico alimentar, somada aos atuais métodos laboratoriais, como a biópsia tecidual ou prova de sobrecarga com frutose. Junto a isso, pode ser realizado o sequenciamento do gene ALDOB, ainda pouco acessível, mas que deve se tornar, em um futuro não tão longínquo, a principal forma de confirmação dessa doença.6 O paciente do caso apresentou, após o contato com frutose, o quadro clínico de hipoglicemia grave (30mg/dl), evoluindo com sonolência e rebaixamento do nível de consciência, necessitando de hospitalização. Após a alta hospitalar, foi orientado ao paciente prosseguir com a investigação, e foram solicitados exames complementares como ultrassom e ressonância magnética de abdome. Estes apresentaram esteatose hepática não alcoólica, caracterizada pelo aumento de gordura hepática, causando o aumento de 5% do volume do órgão, doença bastante prevalente nos pacientes com IHF.9
No caso do paciente do relato, após investigação exaustiva para outras doenças metabólicas que cursassem com hepatopatias na infância, cujos resultados apresentaram-se dentro da normalidade, procedeu-se ao sequenciamento do gene ALDOB, corroborando a hipótese diagnóstica de intolerância hereditária à frutose. Vale ressaltar que uma das mutações em nosso paciente (c.448G>C) foi recentemente observada em pacientes com IHF que desenvolveram infiltração gordurosa hepática, o que talvez implique maior patogenicidade da mesma.9
Curiosamente, os efeitos da ingestão de frutose em indivíduos heterozigotos para IHF (hHFI) foram pouco descritos. Heterozigotos são geralmente considerados possuidores de metabolismo normal da frutose, pois aproximadamente 50% do nível de atividade ALDOB é suficiente para função adequada, e em casos de ingesta excessiva de frutose, podem apresentar leves defeitos metabólicos e maior risco cardiometabólico do que a população normal.8 Os genitores do paciente relatado não apresentaram, entretanto, queixas relacionadas à ingesta de alimentos contendo frutose, mesmo em altas quantidades.
O tratamento das manifestações agudas deve ser realizado buscando a correção da hipoglicemia e da acidose metabólica na perspectiva da prevenção da lesão hepática aguda, como ocorrido no caso descrito.5 O pilar do tratamento no longo prazo consiste na restrição de alimentos ricos em frutose, sacarose e sorbitol, além de medicamentos e suplementos alimentares circulantes em nosso meio. Além disso, há necessidade de suplementos multivitamínicos na prevenção de deficiências de micronutrientes, considerando a dieta restritiva seguida por esses pacientes (Tabela 1).7
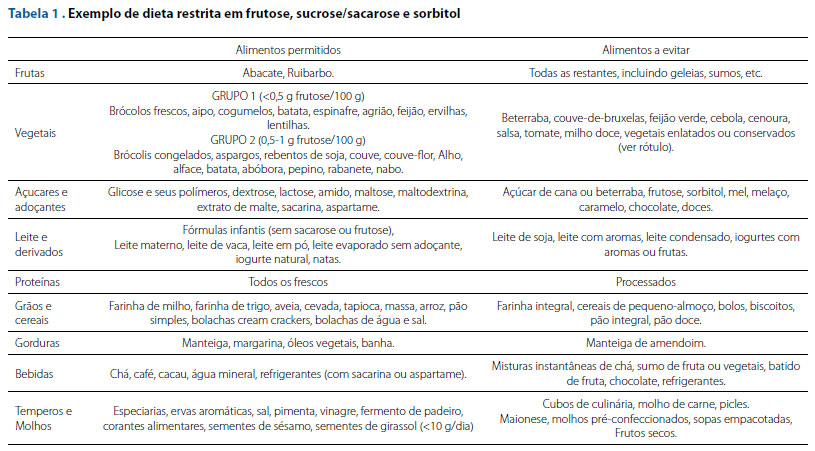
O seguimento da doença dever ser coordenado por uma equipe multiprofissional com conhecimento em patologias metabólicas, além de ser realizado o aconselhamento genético ao doente e sua família, devido ao seu caráter hereditário.9
Mesmo que rara, a IHF deve ser incluída no diagnóstico diferencial de hipoglicemia, sobretudo se ocorrer pós-prandial (hipoglicemia paradoxal). De maneira similar, caso o paciente tenha história de rejeição a elementos que contenham frutose, deve-se também iniciar investigação dirigida para essa suspeita, a fim de se evitar futuras complicações. O diagnóstico realizado a tempo, associado a uma dieta adequada, permite um melhor prognóstico para esses pacientes.
REFERÊNCIAS
1. Tran C. Inborn Errors of Fructose Metabolism. What Can We Learn from Them? Nutrients 2017; 9. doi:10.3390/nu9040356.
2. Demirbas D, Brucker WJ, Berry GT. Inborn Errors of Metabolism with Hepatopathy: Metabolism Defects of Galactose, Fructose, and Tyrosine. Pediatr Clin North Am. 2018;65:337-352.
3. Debray FG, Damjanovic K, Rosset R, et al. Are heterozygous carriers for hereditary fructose intolerance predisposed to metabolic disturbances when exposed to fructose? Am J Clin Nutr. 2018;108(2):292-299
4. Buziau AM, Schalkwijk CG, Stehouwer CDA, Tolan DR, Brouwers MCGJ. Recent advances in the pathogenesis of hereditary fructose intolerance: implications for its treatment and the understanding of fructose-induced non-alcoholic fatty liver disease. Cell Mol Life Sci. 2020;77(9):1709-1719.
5. Li H, Byers HM, Diaz-Kuan A, et al. Acute liver failure in neonates with undiagnosed hereditary fructose intolerance due to exposure from widely available infant formulas. Mol Genet Metab. 2018;123(4):428-432.
6. Di Dato F, Spadarella S, Puoti MG, et al. Daily Fructose Traces Intake and Liver Injury in Children with Hereditary Fructose Intolerance. Nutrients. 2019;11(10):2397.
7. Wilder-Smith CH, Olesen SS, Materna A, Drewes AM. Predictors of response to a low-FODMAP diet in patients with functional gastrointestinal disorders and lactose or fructose intolerance. Aliment Pharmacol Ther. 2017;45(8):1094-1106.
8. Izquierdo-García E, Escobar-Rodríguez I, Moreno-Villares JM, Iglesias-Peinado I. Social and health care needs in patients with hereditary fructose intolerance in Spain. Endocrinol Diabetes Nutr. 2020;67(4):253-262.
9. Aldámiz-Echevarría L, de Las Heras J, Couce ML, et al. Non-alcoholic fatty liver in hereditary fructose intolerance. Clin Nutr. 2020;39(2):455-459.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()