Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(1) - Março 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Síndrome de Aicardi: relato de caso
Aicardi syndrome: a case report
Tifani Dawidowicz Fernandes; Danila de Souza Carraro; Carlos Augusto Takeuchi
DOI:10.31365/issn.2595-1769.v20i1p31-34
Hospital Infantil Sabará, Pediatria - São Paulo - São Paulo - Brasil
Endereço para correspondência:
Recebido em: 19/11/2019
Aprovado em: 09/12/2019
Instituição: Hospital Infantil Sabará, Pediatria
Resumo
INTRODUÇÃO: A síndrome de Aicardi é uma condição genética rara e grave, exclusiva do sexo feminino. Caracterizada por uma tríade composta por: espasmos infantis, lacunas coriorretinianas e agenesia de corpo caloso. Seu diagnóstico é clínico, e sua evolução cursa com crises epiléticas de difícil controle medicamentoso.
OBJETIVO: Relatar a importância de se pensar nessa condição clínica durante a gestação e comprovação da condição clínica logo após o nacimento. Além de demonstrar como fazer o diagnóstico correto e o impacto que o mesmo tem, se realizado precocemente, na preservação da funcionalidade do paciente, buscando um melhor desfecho neurológico.
DESCRIÇÃO DO CASO: Paciente do sexo feminino, com diagnóstico fetal de agenesia de corpo caloso pela ultrassonografia morfológica, apresentou insuficiência respiratória ao nascer, porém, evoluiu bem até os dois meses de idade, quando iniciou quadro de espasmos epilépticos discretos, que evoluíram com a necessidade de medicações anti-epilépticas ao longo dos meses, se tornando uma epilepsia fármaco resistente.
DISCUSSÃO: O diagnóstico da síndrome de Aicardi é difícil de ser realizado, por ser uma síndrome pouco conhecida e também pela própria evolução da doença, que leva algum tempo para ter sua manifestação completa. No entanto, é de extrema importância que seja feito o mais precocemente possível, a fim de evitar complicações e retardar o máximo possível os efeitos neurodegenerativos da doença.
Palavras-chave: Síndrome de Aicardi; Agenesia de corpo caloso; Espasmos Infantis.
Abstract
INTRODUCTION: Aicardi syndrome is a rare and serious genetic condition, exclusively female. Characterized by a triad composed by: infantile spasms, chorioretinal lacunae and corpus callosum agenesis. Its diagnosis is clinical, and its evolution follows epileptic seizures that are difficult to control.
OBJECTIVE: To report the importance of thinking about this clinical condition during pregnancy and proving the clinical condition soon after birth. In addition to demonstrating how to make the correct diagnosis and the impact it has, if performed early, on the preservation of patient functionality, seeking a better neurological outcome.
CASE DESCRIPTION: A female patient with a fetal diagnosis of corpus callosum agenesis by morphological ultrasonography presented respiratory failure at birth. Need for anti-epileptic medications over the months, becoming a drug resistant epilepsy.
DISCUSSION: The diagnosis of Aicardi syndrome is difficult to make because it is a little known syndrome and also due to the evolution of the disease, which takes some time to fully manifest. However, it is of utmost importance that it be done as early as possible in order to avoid complications and delay as much as possible the neurodegenerative effects of the disease.
Keywords: Aicardi Syndrome; Spasms, Infantile; Agenesis of Corpus Callosum.
INTRODUÇÃO
A síndrome de Aicardi (AIC) é uma síndrome genética rara e grave, sem preferência por etnia. Foi relatada por Jean Aicardi e publicada em 1965.1 É uma doença ligada ao cromossomo X, exclusiva do sexo feminino, podendo estar presente apenas no sexo masculino com síndrome de Klinefelter (XXY), enquanto no sexo masculino heterozigoto (46,XY) é letal. No entanto, nenhum gene ou região do cromossomo X foi identificado como causador. 2,3
Classicamente se caracteriza pela tríade composta por: espasmos infantis, lacunas coriorretinianas centrais (patognomônica)3 e agenesia do corpo caloso.4
O diagnóstico da AIC se baseia apenas em características clínicas, pois a etiologia ainda é desconhecida e não há biomarcadores conhecidos associados ao transtorno. No entanto, a AIC é reconhecida como uma desordem pleiotrópica mais complexa, com um espectro expandido de características neurológicas e somáticas. Para incorporar o aumento da complexidade dos recursos, Sutton et al. (2005) propuseram os seguintes critérios diagnósticos modificados:2
• A presença de todas as três características clássicas (tríade clássica) é diagnóstica para AIC.
• A presença de duas características clássicas, além de pelo menos duas características principais ou de suporte, são fortemente sugestivas de um diagnóstico de AIC.
Características principais:
• Malformação cortical (mais comumente a polimicrogiria)
• Heterotopia periventricular e subcortical
• Cistos ao redor do terceiro ventrículo cerebral e/ou plexo coróide
• Coloboma de disco/nervo óptico ou hipoplasia Características de suporte:
• Anormalidades vertebrais e de costela
• Microftalmia
• EEG com padrão espicular "split-brain"
• Assimetria de hemisférios cerebrais
• Malformações vasculares ou malignidade vascular.4
O tratamento é apenas de suporte conforme os sintomas clínicos apresentados pelas meninas.
DESCRIÇÃO DO CASO:
Paciente do sexo feminino, diagnosticada com 23 semanas (2º trimestre) de gestação, pela ultrassonografia morfológica, com malformações do sistema nervoso central, encontradas: agenesia de corpo caloso, cavum do septo pelúcido, colpocefalia e cisto aracnoide. Gestante, foi então encaminhada para exame de cariótipo e microarray por cordocentese, onde foram descartadas alterações cromossômicas, também foi realizada uma ressonância fetal para melhor investigação do quadro, que confirmou a agenesia.
Nascida de parto normal, a termo (38 semanas de idade cronológica), com peso de 3070 gramas ao nascer, estatura de 48 cm, perímetro cefálico 33,3 cm, classificada como adequada para idade gestacional, Apgar 4 no primeiro minuto e 8 no quinto minuto, com desconforto respiratório. Ao exame físico de primeira hora, apresentou as seguintes alterações: leve ptose palpebral direita, nariz em sela e encurtamento da tíbia, restante do exame compatível com a normalidade
Foi levada à UTI neonatal após nascimento devido a insuficiência respiratória aguda com necessidade de oxigênio e para investigação de demais malformações, onde permaneceu 12 dias em investigação neurológica, realizados diversos exames para rastreio de malformações, como:
• Eletroencefalograma (EEG): sem alteração significativa;
• Ultrassonografia transfontanelar: ausência de corpo caloso com assimetria dos ventrículos laterais, com maior dimensão à esquerda;
• Ressonância magnética de crânio que confirmou agenesia de corpo caloso, e observou ainda polimicrogiria, aspecto colpocefálico dos ventrículos laterais, heterotopia da substância cinzenta e
• leucomalácia periventricular (Figura 1);

Figura 1. RNM de crânio: agenesia de corpo caloso, polimicrogiria e leucomalácia periventricular.
• Fundo de olho com lacunas em retina direita e hemorragias retinianas à esquerda (Figura 2);
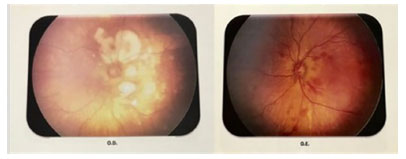
Figura 2. Lacunas à direita e hemorragias retinianas à esquerda.
• Ecodopplercardiograma transtorácico apresentou canal arterial e forame oval patente com hipertensão pulmonar de grau moderado;
• Ultrassonografia de abdome total, rins e vias urinárias: sem alterações.
O quadro descrito acima é sugestivo de Síndrome de Aicardi, porém sem os espasmos característicos da tríade, não foi possível concluir o diagnóstico. Recém-nascida teve alta em bom estado geral, sem necessidade de medicações ou oxigênio domiciliar.
Após receber as vacinas dos 2 meses (pentavalente, poliomielite inativada, pneumocócica 10 valente e rotavírus) iniciou quadro de febre, um pico, e alteração na movimentação ocular de curta duração com perda de consciência e recuperação espontânea. Evoluiu aos longo dos dias, com movimentos ritmados em membro superior direito e inferior esquerdo, de duração fugaz e melhora espontânea. No decorrer de um mês houve nova alteração no padrão e duração das crises, que agora se apresentava com hiperextensão do pescoço e flexão de ambos membros inferiores, de duração mais prolongada. Com isso, foi internada para realizar novo eletroencefalograma que demonstrou espículas e ondas agudas de projeção multifocal, mais frequente no hemisfério direito, com difusão para áreas vizinhas e linha média, provando a presença de atividade epileptiforme, que faltava para completar a tríade clássica de Síndrome de Aicardi.
Foram introduzidos anticonvulsivantes, com controle parcial das crises, que ocorriam principalmente após despertar ou em períodos de sonolência. Apresentou internações recorrentes até os 4 meses, idade atual, para controle das crises por meio de adequação da terapia anti-epilética. Fez uso de carbamazepina, ácido valproico e levetiracetam em doses elevadas, mantendo ainda algumas crises de escape leve, porém com melhora na qualidade de vida. Foi ponderada a necessidade futura de terapia com ACTH (hormônio adrecorticotrópico) e dieta cetogênica para melhor controle da epilepsia.
Com a evolução do quadro, foi possível perceber um atraso global do desenvolvimento neuro- psicomotor e a presença de hemiparesia direita completa e desproporcionada.
DISCUSSÃO
O diagnóstico dessa síndrome se baseia na tríade clássica, no entanto, se houver apenas 2 critérios da tríade, mais dois critérios maiores ou duas características de suporte, se sugere o diagnóstico de síndrome de Aicardi. No caso acima, antes dos espasmos, já haviam critérios clínicos e radiológicos bastante sugestivos de síndrome de Aicardi, porém, o diagnóstico só pode ser confirmado com a tríade clássica, que leva algum tempo para acontecer por completo, gerando ansiedade nos pais e equipe assistencial.
Os pacientes com AIC normalmente iniciam as crises convulsivas aos 3 meses, sendo a maioria antes do primeiro ano de vida; essas vão evoluindo para uma epilepsia grave de padrão diverso. No EEG (eletroencefalograma), normalmente é visualizado um traçado epileptiforme multifocal e assíncrono, com dissociação entre os dois hemisférios.3,4
A síndrome de Aicardi deve ser acompanhada por um neuropediatra, especializado no tratamento de epilepsia, pois o seu tratamento normalmente requer uma combinação de drogas anti- epilépticas para o controle das crises, como visto no caso acima. Esse paciente também deve ter um plano terapêutico individualizado, pois necessita de fisioterapia motora e terapia ocupacional assim que o diagnóstico for realizado, para preservar sua funcionalidade; além de ser interessante também um acompanhamento com a fonoaudiologia e a fisiatria, visando o melhor desenvolvimento neurológico possível.3
A investigação genética não auxilia no diagnóstico dessa síndrome. Pois ainda não temos marcadores moleculares para essa condição. Há um estudo feito por Lund, et al. (2016), onde foram avaliadas 11 meninas com AIC, e se concluiu que a etiologia da AIC não pode ser encontrada no exoma, por não haver uma sequência mutante comum a todos que tem essa síndrome.3,5 A criança também não apresenta fenótipo ou fácies características. O que dificulta o diagnóstico. Apesar de ser uma doença com alteração genética, ela não tem risco aumentado de ser transmitida hereditariamente.2,3
É uma doença grave e de mau prognóstico, com mortalidade elevada, tem sobrevida média de 76% aos 6 anos e 40% aos 14 anos. Sua mortalidade ocorre por infecções pulmonares na maioria dos casos, decorrente do grande comprometimento neurológica que cursa com perda de movimentos, disfagia e da epilepsia refratária.1
A síndrome de Aicardi é uma condição rara, cujo tratamento é apenas de suporte, através do uso de medicações anti-epilépticas para controle das crises e reabilitação. Há dificuldade com relação ao diagnóstico, principalmente devido ao fato de não haver marcador molecular, e a necessidade da tríade para confirmar a síndrome. Futuros estudos com tecnologias melhoradas e novas técnicas de genética molecular poderão fornecer auxílio maior no diagnóstico e tratamento da síndrome.
REFERÊNCIAS BIBLIOGRÁFICAS:
1. Ybazeta, M.A.V.; Chavarría, F.A.T.; Alarcón, N.E.C. Síndrome de Aicardi: Presentación de un caso clínico y revisión de la literatura. Rev Neuropsiquiatria 2016, 79 (1): 59-65. Available from: http://www.scielo.org.pe/scielo.phpscript=sci_arttext&pid=S0034-85972016000100008&lng=es.,
2. B.K.Y; Sutton, V.R.. Aicardi syndrome, an unsolved mystery: Review of diagnostic features, previous attempts, and future opportunities for genetic examination. American Journal of Medical Genetics Part C. 2018; 178C: 423-431. Available from: http://doi.org/10.1002/ajmg.c.31658
3. Sutton VR, Van den Veyver IB. Aicardi Syndrome. 2006 Jun 30 [Updated 2014 Nov 6].GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2019. Available from: http://www.ncbi.nlm.nih.gov/books/NBK1381/
4. Govil-Dalela T, Kumar A, Agarwal R, Chugani HT. Agenesis of the Corpus Callosum and Aicardi Syndrome: A Neuroimaging and Clinical Comparison. Pediatr Neurol. 2017
5. Mar;68:44-48.e2. doi: 10.1016/j.pediatrneurol.2016.12.002. Epub 2017 Jan 4
6. Lund C, Striano P, Sorte H, S, Parisi P, Iacomino M, Sheng Y, Vigeland M, D, Øye A, -M, Steensbjerre Møller R, Selmer K, K, Zara F: Exome Sequencing Fails to Identify the Genetic Cause of Aicardi Syndrome. Molecular Syndromology 2016;7:234-238. Doi:10.1159/000448367
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()