Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(1) - Março 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Artigo de Revisao
Pediatra/Neonatologista: o primeiro médico na avaliação de crianças com ambiguidade genital
Pediatrician/Neonatologist: the first doctor in the evaluation of children with sex ambiguity
Andrea Trevas Maciel-Guerra1,2; Gil Guerra-Junior2,3
DOI:10.31365/issn.2595-1769.v20i1p18-25
1. Faculdade de Ciências Médicas da Universidade Estadual de Campinas (Unicamp), Departamento de Genética Médica e Medicina Genomica - Campinas - SP - Brasil
2. Faculdade de Ciências Médicas da Universidade Estadual de Campinas (Unicamp), Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo (GIEDDS) - Campinas - SP - Brasil
3. Faculdade de Ciências Médicas da Universidade Estadual de Campinas (Unicamp), Departamento de Pediatria - Campinas - SP - Brasil
Endereço para correspondência:
Recebido em: 21/01/2020
Aprovado em: 22/01/2020
Instituição: Universidade Estadual de Campinas (Unicamp)
Resumo
OBJETIVO: Apresentar os conhecimentos fundamentais de que o pediatra ou neonatologista deve dispor para avaliar crianças com ambiguidade genital (AG) e orientar os familiares.
MÉTODOS: Revisão de literatura científica por meio de artigos publicados no PubMed, destacando-se o Consenso de Chicago publicado em 2006 e sua revisão em 2016.
RESULTADOS: O pediatra ou neonatologista deve: (1) conhecer a diferenciação sexual normal; (2) saber identificar uma AG e qual criança deve ser investigada; (3) obter os principais dados de história clínica e exame físico; (4) saber quais os exames laboratoriais iniciais; (5) conhecer as principais etiologias das AG; (6) saber conversar com a família sobre o sexo de criação.
CONCLUSÕES: Apesar dos casos de AG serem relativamente raros na população e pouco observados na prática diária do pediatra, seu diagnóstico precoce e correto requer atenção médica adequada, possibilitando o estabelecimento do prognóstico (puberdade, fertilidade e risco de neoplasia), o planejamento terapêutico e o aconselhamento genético, reduzindo a ansiedade da família e o risco de problemas psicológicos e sociais. A equipe multidisciplinar é fundamental para o sucesso diagnóstico e terapêutico. O pediatra ou neonatologista é peça chave nessa equipe, pois será o responsável pelas primeiras informações dadas à família e deverá assumir o papel de interlocutor entre esta e a equipe, respeitando suas particularidades sociais, culturais, econômicas e religiosas.
Palavras-chave: Doenças dos Genitais Masculinos; Identidade de Gênero; Hipospadia.
Abstract
OBJECTIVE: To present the main knowledge that the pediatrician or neonatology must have to evaluate children with genital ambiguity (GA) and provide guidance to family members.
METHODS: Review of scientific literature through articles published in PubMed, highlighting the Chicago Consensus published in 2006 and its review in 2016.
RESULTS: The pediatrician or neonatologist should: (1) know normal sexual differentiation; (2) know how to identify a GA and which child should be investigated; (3) look for key data in clinical history and physical examination; (4) know the first laboratory tests to be asked; (5) know the main etiologies of GA; (6) know how to talk to the family about sex of rearing.
CONCLUSIONS: Though cases of GA are relatively rare in the population and rarely observed in daily pediatric practice, early and correct diagnosis requires adequate medical attention, in order to allow the establishment of prognosis (puberty, fertility and risk of neoplasia), therapeutic planning and genetic counseling, reducing family anxiety and the risk of psychological and social problems. The multidisciplinary team is essential for diagnostic and therapeutic success. The pediatrician or neonatologist is a key player in this team, being responsible for the first information given to the family, and should assume the role of interlocutor between the family and the team, respecting their social, cultural, economic and religious particularities.
Keywords: Gender Identity; Male Urogenital Diseases; Hypospadias.
INTRODUÇÃO
Desde o Consenso de Chicago, publicado em 20061, os Distúrbios (ou Diferenças) da Diferenciação do Sexo (DDS) são definidos como anomalias congênitas nas quais o desenvolvimento do sexo cromossômico, gonadal ou anatômico é atípico1. Os DDS se manifestam por ambiguidade ou atipia genital (AG); por atraso puberal (em geral com hipogonadismo hipergonadotrófico); puberdade heterossexual (com virilização em meninas ou feminilização em meninos); ou ainda por infertilidade1,2. São em geral afecções raras, com uma incidência média estimada de um em cada 4.000 a 5.000 nascimentos vivos2.
Frente a uma criança com AG, o pediatra ou neonatologista deve ter em mente a importância de seu papel em uma situação ainda cercada de preconceitos e acompanhada de implicações médicas, psicológicas e sociais. De fato, o manejo dos DDS exige muita sensibilidade, de modo que ao longo do tempo não exista confusão a respeito da identificação sexual da criança, do adolescente ou do indivíduo adulto1-5.
O grande desafio frente a pacientes com DDS, principalmente os recém-nascidos com AG, é estabelecer o diagnóstico etiológico preciso no menor tempo possível. Deste diagnóstico depende não só a definição do sexo, mas também todos os procedimentos terapêuticos subsequentes e, ainda, o aconselhamento genético da família1-5.
Em todos os casos, é fundamental que o diagnóstico seja feito antes do estabelecimento da identidade sexual, idealmente ainda no período neonatal1-6.
É necessário o envolvimento de vários profissionais da área da saúde - pediatra (neonatologista), endocrinologista, geneticista, cirurgião, ginecologista, radiologista, anatomopatologista, médico-legista, psicólogo (ou psiquiatra) e assistente social. A atuação conjunta e integrada desses profissionais permite não só maior rapidez no diagnóstico, mas também a uniformização das informações transmitidas à família e, consequentemente, maior confiança da família na equipe médica como um todo. Além disso, a concentração destes casos em serviços terciários, como hospitais universitários, possibilita contato entre pacientes e famílias com experiências semelhantes1-4,7.
Ao identificar um recém-nascido com AG, a atuação do "primeiro médico" - pediatra ou neonatologista - na abordagem da família é fundamental, já que sua palavra pode ser tomada como "verdade absoluta" e dificilmente será desfeita, mesmo se não estiver correta. Ao informar com tranquilidade que não há como definir de imediato o sexo da criança, e que esta definição depende de investigação laboratorial minuciosa, ajuda a evitar algumas das sequelas que o problema pode acarretar1-4. É fundamental que esse médico garanta à família que a criança tem um sexo, porém o mesmo não pode ser definido exclusivamente com a inspeção dos genitais. Nunca deve ser dito que a criança tem dois sexos ou não tem sexo nenhum; isso é um erro grave que deve ser evitado1-4.
Quando a identificação da AG ocorre logo ao nascimento, a família deve ser imediatamente comunicada da alteração genital, da necessidade da realização de exames especializados e de avaliação por profissionais experientes (geralmente atuantes em hospitais universitários) e, portanto, do atraso no registro civil1-7.
São seis as informações que todo pediatra ou neonatologista devem ter para conversar com as famílias de crianças com DDS e AG.
1ª INFORMAÇÃO: CONHECER A DIFERENCIAÇÃO SEXUAL NORMAL8-11
Para explicar à família a causa da AG, o pediatra (ou outro profissional da saúde) precisa entender as influências genéticas e hormonais responsáveis pelo desenvolvimento normal das gônadas, dutos genitais internos e genitália externa.
Até cerca de seis a sete semanas após a fertilização, o embrião humano é um organismo bissexual, equipado com primórdios gonadais e genitais idênticos nos dois sexos, e não é possível distinguir macro ou microscopicamente embriões com predestinação masculina ou feminina. Esse estado sexualmente neutro é representado por rudimentos gonadais (gônadas bipotentes), primórdios dos condutos genitais internos masculinos e femininos (dutos de Wolff e Müller, respectivamente) e rudimentos genitais externos (tubérculo genital, pregas genitais, saliências labioescrotais e seio urogenital).
Dependendo do sexo genético do embrião (46,XY ou 46,XX) e da expressão de uma série de genes ocorrerá a formação de testículos ou ovários, processo conhecido como determinação sexual. A diferenciação sexual, por sua vez, diz respeito aos processos subsequentes à formação das gônadas, ou seja, referente ao desenvolvimento dos genitais internos e externos.
Em embriões de sexo genético masculino (46,XY e com expressão normal do gene SRY - localizado no braço curto do cromossomo Y - e dos demais genes envolvidos na determinação testicular), a formação do testículo se inicia por volta da 7ª semana. As células de Sertoli se diferenciam e se agrupam formando cordões que englobam as células sexuais primitivas - que se tornam, assim, espermatogônias. Esses cordões desenvolvem-se para formar túbulos seminíferos, túbulos retos e rede testis. As células de Leydig podem ser observadas entre os túbulos a partir da oitava semana.
Uma vez diferenciado o testículo, este é responsável por conduzir tanto a regressão dos primórdios dos genitais internos femininos quanto a diferenciação dos dutos internos e genitais externos masculinos. A partir da 7ª semana, as células de Sertoli secretam o hormônio anti-mülleriano (HAM), que induz a regressão dos dutos de Müller. A ação do HAM ocorre por via parácrina, de modo que cada testículo é responsável pela destruição do duto de Müller de seu lado12. Entre a 8ª e a 9ª semanas, as células de Leydig secretam testosterona, que estabiliza os dutos de Wolff e permite sua diferenciação em epidídimos, canais deferentes, vesículas seminais e duto ejaculatório. A ação local da testosterona sobre os dutos de Wolff é muito mais importante que sua ação sistêmica, de modo que cada testículo também é responsável pela diferenciação do duto de Wolff de seu lado13.
A testosterona é convertida pela enzima 5α-redutase tipo 2 em dihidrotestosterona (DHT), andrógeno mais potente que viriliza os rudimentos genitais externos entre a 9ª e a 12ª semanas de gestação. Pela ação desse hormônio, o tubérculo genital dá origem à glande e aos corpos cavernosos; as pregas genitais alongam-se juntamente com o tubérculo e fundem-se para formar o corpo esponjoso; ocorre fusão das saliências labioescrotais na linha média, originando a bolsa escrotal; e o seio urogenital dá origem à uretra peniana. Na glande, uma invaginação ectodérmica forma a porção balânica da uretra, que se une à porção peniana por volta da 12ª semana; o prepúcio envolve quase por completo a glande em torno da 14ª semana.
A migração dos testículos da cavidade pélvica para a bolsa escrotal inicia-se por volta da 28ª semana, completando-se, em geral, em torno da 32ª. A gonadotrofina coriônica humana (hCG), produzida pelo sinciotrofoblasto, estimula a secreção de testosterona pelas células de Leydig durante o período crítico da diferenciação sexual masculina, ou seja, a primeira metade da gestação. A partir de então, o hormônio luteinizante (LH) do próprio feto é necessário para a continuidade do estímulo das células de Leydig, de modo a promover a completa descida testicular e o crescimento peniano.
Em embriões de sexo genético feminino (46,XX - portanto com ausência de SRY - e na presença de genes necessários para a manutenção da estrutura e função ovarianas) as gônadas permanecem no estado bipotente até o final da 10ª semana, quando se inicia a determinação dos ovários. Para a manutenção ovariana é necessária a presença de dois cromossomos X íntegros, caso contrário acelera-se o processo de degeneração dos folículos ovarianos e a gônada torna-se disgenética, ou seja, constituída somente de tecido conjuntivo, sem elementos da linhagem germinativa. Uma vez que não há secreção de HAM, os dutos de Müller se desenvolvem para formar o trato genital feminino (útero, trompas e porção superior da vagina). Na ausência de altas concentrações locais de testosterona, não há diferenciação dos dutos de Wolff, que persistem como resquícios embrionários. Na falta de estímulo pela DHT, o tubérculo genital dá origem à glande e à haste do clitóris, as pregas genitais aos pequenos lábios, as saliências labioescrotais aos grandes lábios, e o seio urogenital divide-se para formar a uretra feminina e a porção inferior da vagina.
2ª INFORMAÇÃO: O QUE É AG? QUEM DEVE SER INVESTIGADO?
Nem sempre o médico dá a devida atenção ao exame da genitália do recém-nascido, e em vários casos o achado de uma AG é feito por um membro da família. É importante o conhecimento das variações da normalidade que, para o profissional menos experiente, podem se confundir com AG. É o caso de prepúcio mais desenvolvido no clitóris sem tecido cavernoso, que não configura hipertrofia do clitóris, ou de excesso de adiposidade em região pubiana, que pode dar a falsa impressão de micropênis. Por outro lado, a não palpação de gônadas numa genitália de aspecto masculino, frequentemente considerada apenas como criptorquidia bilateral, pode ser o modo de apresentação de meninas com Hiperplasia Adrenal Congênita (HAC) com grau extremo de virilização intrauterina1,2. A associação de criptorquidia e hipospadia deve alertar ainda mais o pediatra.
Os critérios diagnósticos propostos por Danish14 em 1982 são os mais citados na literatura e facilmente aplicáveis à prática médica. De acordo com esse autor, a AG existe em qualquer uma das características listadas a seguir.
Em genitália de aparente aspecto masculino: (1) gônadas não palpáveis; (2) tamanho peniano esticado abaixo de -2,5 desviospadrão em relação à média para a idade (Tabela 1)15; (3) gônadas pequenas, ou seja, maior diâmetro inferior a 8 mm; (4) presença de massa inguinal (que poderá corresponder a útero e trompas); (5) hipospadia médio-peniana a perineal.
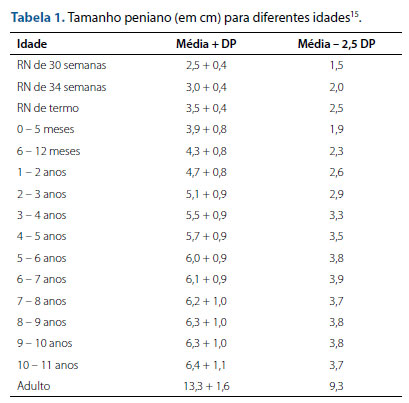
Em genitália de aparente aspecto feminino: (1) diâmetro clitoriano superior a 6 mm (ou clitóris visível e palpável com as pernas totalmente aduzidas/fechadas); (2) gônada palpável em saliência labioescrotal; (3) fusão labial posterior; (4) massa inguinal que possa corresponder a testículos.
Não se fixando em critérios rígidos, pode-se dizer que existe AG sempre que houver dificuldade para que o médico, supostamente conhecedor das variantes da normalidade de uma genitália externa, possa atribuir o sexo à criança. É fundamental, porém, diferenciar AG de malformação genital, como ocorre na epispadia, na inversão penoescrotal parcial ou total, e na agenesia de pênis ou clitóris, que não devem ser consideradas como DDS1,2.
3ª INFORMAÇÃO: A IMPORTÂNCIA DA HISTÓRIA CLÍNICA E DO EXAME FÍSICO
Na anamnese devem ser avaliados os antecedentes gestacionais, com especial atenção ao uso de medicamentos e aos sinais de virilização materna; história de baixo peso ao nascimento16; antecedentes familiais, como consanguinidade entre os pais, casos semelhantes, história familial de atraso ou avanço puberal, infertilidade, hipertensão arterial na infância ou mortes inexplicadas nos primeiros meses de vida, entre outros1-7.
Quanto ao exame físico, deve-se ter em mente que "os achados no exame dos genitais não definem o diagnóstico etiológico, mas podem priorizar a realização de exames". Deve-se procurar por dismorfismos ou malformações (em especial de coluna e anorretais) que configurem um quadro sindrômico e avaliar os genitais externos. Na criança maior, é importante avaliar também o estado nutricional, pressão arterial, presença de pêlos sexuais, acne e sinais puberais. A genitália externa deve ser avaliada determinando o grau de virilização (Figura 1 e Quadro 1) com base na análise do tamanho do falo; da posição do meato uretral; da presença de intróito vaginal ou abertura de seio urogenital; do grau de fusão, simetria, pigmentação e enrugamento das saliências labioescrotais; e da presença de massas inguinais, assim como a localização e tamanho das gônadas1-7.
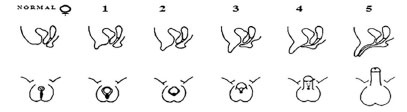
Figura 1. Grau 1: Genitália de aspecto feminino, com somente aumento do falo; Grau 2: maior aumento do falo, associado à fusão posterior das saliências labioescrotais, sem seio urogenital; Grau 3: importante aumento do falo, associado à fusão quase completa das saliências labioescrotais, e presença de seio urogenital com abertura perineal; Grau 4: falo de aspecto peniano, associado à fusão completa das saliências labioescrotais, e presença de seio urogenital com abertura perineal na base do falo ou em qualquer lugar do corpo do falo, exceto a glande; Grau 5: falo de aspecto peniano, associado a fusão completa das saliências labioescrotais, e presença de seio urogenital com abertura balcânica (no caso de cariótipo 46,XX sem gônadas palpáveis; no caso de cariótipo 46,XY com micropênis com ou sem gônadas palpáveis) .

A classificação dos graus de virilização da genitália externa de 1 a 5 proposta por Prader17 para meninas virilizadas com HAC também é de grande utilidade para descrição de outras causas de DDS. Mais recentemente, Ahmed e cols. desenvolveram o Escore de Masculinização da Genitália Externa (EMS - External Masculinization Score)18 utilizado apenas para indivíduos com cromossomo Y no cariótipo e que leva em consideração o tamanho peniano, a fusão labioescrotal, e a posição do meato uretral e de cada uma das gônadas ( Quadro 1).
4ª INFORMAÇÃO: OS EXAMES LABORATORIAIS INICIAIS
Embora a definição do sexo genético não seja suficiente para que se tome decisões acerca do sexo de criação, sua realização é fundamental para direcionar a investigação laboratorial. O exame do cariótipo permite ainda detectar alterações numéricas ou estruturais em cromossomos sexuais ou autossomos, em cariótipos homogêneos ou em mosaico (mais de uma linhagem de células somáticas em um mesmo indivíduo oriundas da mesma fonte genética, como 45,X/46,XY), e também a presença de quimerismo (mais de uma linhagem de células somáticas em um mesmo indivíduo oriundas de fontes genéticas diferentes, como 46,XX/46,XY). As anomalias numéricas ou estruturais dos cromossomos sexuais e o quimerismo 46,XX/46,XY determinam anomalias da determinação gonadal; assim sendo, sua detecção indica a necessidade de realizar biópsia gonadal para definição diagnóstica. Por sua vez, anomalias autossômicas são mais frequentemente observadas em quadros de anomalias congênitas múltiplas associadas a deficiência de crescimento e de desenvolvimento neuropsicomotor, caracterizando quadros sindrômicos1-7.
Nos casos de AG sem gônadas palpáveis a hipótese de HAC deve ser investigada mesmo antes do resultado do cariótipo, por ser esta a principal causa de AG em recém-nascidos e pelo fato da forma perdedora de sal ser potencialmente letal. Esta geralmente se manifesta por hiponatremia, hipercalemia, acidose metabólica e hipovolemia a partir da 2ª ou 3ª semana de vida (ou até mais tarde), quando a criança já está em casa. Como a principal causa de HAC é a deficiência da 21-hidroxilase, ao menos a mensuração da 17-OH-progesterona sérica deve ser realizada nesses casos (ver resultado do teste de triagem neonatal), e, quando possível, realizar também a avaliação frequente (diária ou a cada dois dias) de sinais clínicos, do peso e das dosagens séricas de sódio e potássio1-7,.
Os exames hormonais (as dosagens basais de LH, FSH, testosterona e DHT só têm valor quando realizadas na minipuberdade, entre 30 e 100 dias de vida, ou na puberdade) e o cariótipo devem ser realizados em serviços especializados, assim como a avaliação por imagem de útero, gônadas, próstata e seio urogenital, que é necessária, porém nem sempre conclusiva1-7,19. Os procedimentos mais utilizados são ultrassonografia em associação com genitografia (ou uretrocistografia miccional retrógrada), e, menos frequentemente, tomografia computadorizada ou ressonância nuclear magnética da região pélvica20. A laparoscopia vem sendo realizada com maior frequência e traz maior precisão a essa avaliação; a ela deve se associar, sempre que possível, cistoscopia intraoperatória, que fornece informações importantes a respeito da presença ou não de seio urogenital20.
5ª INFORMAÇÃO: PRINCIPAIS ETIOLOGIAS DE DDS COM AG
São várias as classificações dos DDS encontradas na literatura, uma vez que os critérios para agrupar as diversas anomalias são bastante heterogêneos21,22. Como consequência da enorme complexidade do assunto, todas elas podem ser questionadas em alguns de seus aspectos. Até mesmo a do Consenso de Chicago1, que não conseguiu eliminar totalmente problemas em relação à terminologia e não permitiu afastar totalmente algum grau de estigmatização, como no caso de DDS ovotesticular (antigo hermafroditismo verdadeiro). Em primeiro lugar, a sugestão de se incluir o cariótipo no nome da doença supõe, erroneamente, que os pacientes não tenham conhecimento do que significa ser 46,XY ou 46,XX. Embora a discussão a respeito da nomenclatura seja bem-vinda, devese continuar na busca de termos alternativos que sejam realmente neutros e não tragam, em si, a conotação de um sexo que pode não condizer com o escolhido para aquele paciente em particular. De todo modo, a sugestão feita pelo Consenso de Chicago de substituir o termo intersexo por DDS teve uma acolhida unânime2. A seguir estão listadas algumas das mais frequentes etiologias das DDS1,2,21,22.
A forma clássica da HAC por deficiência da enzima 21-hidroxilase (DDS - 46,XX - fetal - 21- hidroxilase) é uma das principais causas de DDS e a mais frequente causa de virilização de fetos de sexo genético feminino (46,XX), correspondendo a 80 a 90% dos casos. Pode se manifestar sob duas formas clínicas, virilizante simples e perdedora de sal. A forma virilizante simples corresponde a 20 a 30% dos casos e determina AG em recém-nascidos do sexo feminino; quando não tratada acarreta, em ambos os sexos, virilização pós-natal progressiva, com sinais e sintomas evidentes de pseudo-puberdade precoce (aumento do clitóris, aumento do pênis sem correspondente aumento testicular, pubarca, hirsutismo, acne, engrossamento da voz, avanço da velocidade de crescimento e da maturação esquelética). Na forma perdedora de sal, que corresponde a 70 a 80% dos casos, as manifestações clínicas incluem, além do quadro de virilização pré-natal no sexo feminino e pós-natal em ambos os sexos, desde formas graves de desidratação hiponatrêmica e hipercalêmica, vômitos, acidose metabólica, choque hipovolêmico e óbito, se não houver tratamento adequado, até quadros mais discretos, onde somente são observados baixo ganho ponderal, alterações de eletrólitos e dosagem aumentada da renina. Nesses casos há risco de desidratação e choque quando as crianças são submetidas a situações de estresse sem o adequado tratamento glico e mineralocorticóide.
Dentre as causas de AG em fetos de sexo genético masculino (46,XY) destacam-se a Insensibilidade Parcial aos Andrógenos (DDS - 46,XY - Insensibilidade Parcial aos Andrógenos) e a deficiência da enzima 5α-redutase tipo 2 (DDS - 46,XY - Deficiência da 5α-redutase tipo 2), que ao nascimento se manifestam por graus variados de micropênis, hipospadia e criptorquidia, sendo, portanto, clinicamente indistinguíveis entre si, bem como de outras causas de deficiência de virilização de fetos masculinos. O diagnóstico definitivo frequentemente depende de extensa avaliação laboratorial, incluindo avaliação molecular dos genes AR (Androgen Receptor) e SRD5A2 (Steroid 5-Alpha-Reductase type 2). Na puberdade, a Insensibilidade Parcial aos Andrógenos (herança recessiva ligada ao X) caracteriza-se por ginecomastia e pouca virilização genital e pilificação corpórea, enquanto que na deficiência de 5α- redutase tipo 2 (herança autossômica recessiva) há virilização genital, embora nem sempre com aumento peniano adequado, ausência de ginecomastia e hipoplasia ou ausência de próstata.
Finalmente, dentre as causas de AG associadas aos distúrbios da determinação gonadal, destacam- se a Disgenesia Gonadal Parcial (DDS - 46,XY - Disgenesia Gonadal Parcial), a Disgenesia Gonadal Mista (DDS por anomalia de cromossomos sexuais - cariótipo 45,X/46,XY - Diagenesia Gonadal Mista) e o DDS ovotesticular (DDS XX ou XY ou por anomalia de cromossomos sexuais - DDS ovotesticular). A Disgenesia Gonadal Parcial é caracterizada pela presença de cariótipo 46,XY, sem mosaicismo, em indivíduos com diferenciação testicular parcial ou gônadas disgenéticas, evidência de derivados dos dutos de Müller e AG sem sinais clínicos da síndrome de Turner. Na Disgenesia Gonadal Mista, o quadro fenotípico é semelhante ao da Disgenesia Gonadal Parcial, porém com sinais da síndrome de Turner e cariótipo com mosaico 45,X/46,XY. O DDS ovotesticular é também um diagnóstico histológico; caracteriza-se pela presença de tecido ovariano (com folículos) e testicular (com túbulos seminíferos, com ou sem espermatozóides) no mesmo indivíduo, em uma mesma gônada (denominada ovotestis) ou em gônadas opostas.
6ª INFORMAÇÃO: O SEXO DE CRIAÇÃO
Na grande maioria das vezes, o diagnóstico do sexo é realizado ao nascimento de forma correta e sem qualquer dificuldade, com base apenas nas características da genitália externa. Em situações normais, há concordância entre genitais externos (sexo genital externo) e internos (sexo genital interno), gônadas (sexo gonadal) e sexo cromossômico. A produção hormonal (sexo endocrinológico) tem papel fundamental não só a partir da puberdade, quando os indivíduos desenvolvem caracteres sexuais secundários e capacidade reprodutiva, mas também durante o desenvolvimento fetal. Finalmente, a concordância entre os sexos cromossômico, gonadal, genital interno, genital externo e endocrinológico pode ficar prejudicada se não houver a correspondente identificação psicológica do indivíduo (sexo psicológico), também devendo levar em conta sua inserção social em um ou outro sexo (sexo social)1,2.
Entretanto, em situações patológicas, o sexo de criação somente pode ser definido se forem levados em conta outros dados. Frente a uma criança com AG, o objetivo principal é o diagnóstico preciso da sua etiologia, o que permitirá a correta definição do sexo1,2, a estimativa do risco de malignização gonadal e da época adequada da gonadectomia (quando indicada)2,23,24, a definição da época e do tipo de correção cirúrgica reconstrutiva da genitália1,2, a previsão quanto ao desenvolvimento de caracteres sexuais secundários espontâneos1,2, a necessidade de terapia de reposição ou substituição, a possibilidade de fertilidade futura1,2 e, finalmente, o aconselhamento genético e o acompanhamento psicológico da família e do paciente1,2.
Há evidências de que a definição do sexo de criação e aceitação da sexualidade diferem significativamente entre várias regiões, quer por aspectos sociais, culturais ou religiosos. Na maioria das sociedades a posição social e econômica dos homens difere da alcançada pelas mulheres de forma significativa, e o sexo masculino parece oferecer nesses casos mais e melhores opções de vida. A bagagem cultural do próprio médico pode também influenciar de alguma forma na decisão do sexo de criação, o que reforça a importância da equipe interdisciplinar25. Portanto, na discussão com a família para a tomada da decisão do sexo de criação, não se deve deixar de ponderar sobre aspectos sociais, culturais, étnicos e religiosos próprios daquela família ou da sociedade onde ela está inserida1,2,25.
Do ponto de vista legal, no Brasil, o Conselho Federal de Medicina (Resolução nº 1664 de maio de 2003) estabelece no Artigo 2 que "Pacientes com DDS devem ter assegurada uma conduta de investigação precoce com vistas a uma definição adequada do gênero e tratamento em tempo hábil". Ainda de acordo com o Conselho Federal de Medicina, no Artigo 4, "é necessária uma estrutura mínima que permita a realização de exames hormonais, genéticos, de imagem e de patologia. Para a definição final e adoção do sexo de criação é obrigatória a existência de equipe multiprofissional, que assegure os conhecimentos nas áreas de pediatria, endocrinologia pediátrica, endocrinologia, genética, psiquiatria infantil e cirurgia". É uma exigência legal, portanto, que o ambiente de avaliação garanta segurança e suporte no acompanhamento. No entanto, não são todos os hospitais que contam com profissionais com experiência nessa área26.
A família tem todo o direito à assistência médica e jurídica, apoio e informações sobre o problema e suas consequências durante todo o processo de avaliação. O paciente, dependendo da idade e condições de entendimento, também pode participar da definição de seu próprio sexo. Está determinado pelo Conselho Federal de Medicina que "no momento da definição final do sexo, os familiares ou responsáveis legais, e, eventualmente, o paciente, devem estar devidamente informados de modo a participar da decisão do tratamento proposto"26.
Quando a revisão de registro de nome e sexo legal é solicitada para crianças que já possuem uma identidade psicológica e social ou para adolescentes, esse processo requer avaliação profunda e detalhada, feita por vários profissionais. É fundamental a participação da família e, quando possível, do próprio interessado (o paciente), na decisão do sexo de criação e na programação terapêutica, em especial, a época da(s) cirurgia(s) corretiva(s). Esse é geralmente um processo longo e que exige muita colaboração do paciente e dedicação da equipe médica. Considera-se hoje que alguns registros podem ser modificados desde que essa mudança seja feita para a real integração social e psicológica dos pacientes26.
É importante salientar que, nos últimos anos, um movimento liderado por sociedades de pacientes com DDS tem combatido fortemente a prática realizada por alguns médicos ou equipes de tomar decisões unilaterais a respeito da definição do sexo de criação e do tratamento da AG, em especial quando estas decisões foram realizadas com informações incompletas às famílias e as correções cirúrgicas genitais foram precoces e definitivas. No contexto ético atual, esta decisão tem que ser totalmente compartilhada com os responsáveis pelo paciente ou com ele próprio, quando possível. A família deve receber todas as informações e ter o tempo que for necessário para participar da definição junto com a equipe médica do sexo de criação e do planejamento cirúrgico de correção dos genitais1,2,27-30.
CONSIDERAÇÕES FINAIS
Apesar dos DDS com AG serem relativamente raros na população e pouco observados na prática diária do pediatra, seu diagnóstico precoce e correto requer atenção médica adequada, de modo a possibilitar o estabelecimento do prognóstico (puberdade, fertilidade e risco de neoplasia), o planejamento terapêutico e o aconselhamento genético, reduzindo de forma acentuada a ansiedade da família e o risco de problemas psicológicos e sociais. Uma equipe multi ou interdisciplinar atuando em serviço terciário é fundamental para o sucesso diagnóstico e terapêutico. O pediatra é peça chave nessa equipe, pois frequentemente será o responsável pelas primeiras informações dadas à família e deverá assumir o papel de interlocutor entre esta e a equipe multi ou interdisciplinar, respeitando suas particularidades sociais, culturais, econômicas e religiosas.
AGRADECIMENTOS
A todos aqueles que direta ou indiretamente têm contribuído para o crescimento do Grupo Interdisciplinar de Estudos da Determinação e Diferenciação do Sexo (GIEDDS) da FCM - UNICAMP, como os Drs. Antonia Paula Marques-de-Faria, Juliana Gabriel Ribeiro de Andrade, Maricilda Palandi de Mello, Roberto Benedito de Paiva-e-Silva e Márcio Lopes Miranda, os residentes, os alunos de graduação e pós-graduação, os estagiários, os pacientes e as famílias.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Lee PA, Houk CP, Ahmed SF, Hughes IA; International Consensus Conference on Intersex organized by the Lawson Wilkins Pediatric Endocrine Society and the European Society for Paediatric Endocrinology. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118:488-500.
2. Lee PA, Nordenström A, Houk CP, Ahmed SF, Auchus R, Baratz A, et al. Global Disorders of Sex Development Update since 2006: Perceptions, Approach and Care. Horm Res Paediatr. 2016;85(3):158-80.
3. American Academy of Pediatrics. Evaluation of the newborn with developmental nomalies of the external genitália. Pediatrics. 2000;106:138-42.
4. Guerra-Júnior G, Maciel-Guerra AT. The role of the pediatrician in the management of children with genital ambiguities. J Pediatr (Rio J). 2007;83(5 Suppl):S184-91.
5. Ogilvy-Stuart AL, Brain CE. Early assessment of ambiguous genitalia. Arch Dis Child. 2004;89:401-7.
6. Nelson CP, Gearhart JP. Current views on evaluation, management, and gender assignment of the intersex infant. Nat Clin Pract Urol. 2004;1:38-43.
7. Eugster EA, Hains D, DiMeglio LA. Initial management of infants with intersex conditions in a tertiary care center: a cautionary tale. J Pediatr Endocrinol Metab. 2006;19:191-2.
8. Migeon CJ, Wisniewski AB. Human sex differentiation and its abnormalities. Best Pract Res Clin Obstet Gynaecol. 2003;17:1-18.
9. Hughes IA. Minireview: sex differentiation. Endocrinology. 2001;142:3281-7.
10. Guerra-Junior G, Maciel-Guerra AT. A determinação e diferenciação sexual normais: atualização. Arq Bras Encocrinol Metab. 1997;41:191-7.
11. Moraes SG, Maciel-Guerra AT. Aspectos embriológicos. In: Menino ou menina? Os Distúrbios da Diferenciação do Sexo. Curitiba: Editora Appris, 2019. pp. 21-34.
12. MacLaughlin DT, Donahoe PK. Mullerian inhibiting substance: an update. Adv Exp Med Biol. 2002;511:25-38.
13. Hannema SE, Hughes IA. Regulation of Wolffian duct development. Horm Res. 2007;67:142- 51.
14. Danih RK. Intersex problems in the neonate. Indian J Pediatr. 1982;49:555-75.
15. Gabrich PN, Vasconcelos JS, Damião R, Silva EA. Penile anthropometry in Brazilian children and adolescents. J Pediatr (Rio J). 2007;83(5):441-6.
16. Machado-Neto FA, Morcillo AM, Maciel-Guerra AT, Guerra-Junior G. Idiopathic male pseudohermaphroditism is associated with prenatal growth retardation. Eur J Pediatr. 2005;164:287-91.
17. Prader A. Genital findings in the female pseudo-hermaphroditism of the congenital adrenogenital syndrome; morphology, frequency, development and heredity of the different genital forms. Helv Paediatr Acta. 1954;9:231-48.
18. Ahmed SF, Khwaja O, Hughes IA. The role of a clinical score in the assessment of ambiguous genitalia. BJU Int. 2000;85(1):120-4.
19. Bertelloni S, Russo G, Baroncelli GI. Human chorionic gonadotropin test: old uncertainties, new perspectives, and value in 46,XY Disorders of Sex Development. Sex Dev. 2018;12(1- 3):41-9.
20. Guerra-Junior G, Andrade KC, Barcelos IHK, Maciel-Guerra AT. Imaging techniques in the diagnostic journey of Disorders of Sex Development. Sex Dev. 2018;12(1-3):95-9.
21. Cox K, Bryce J, Jiang J, Rodie M, Sinnott R, Alkhawari M, et al. Novel associations in disorders of sex development: findings from the I-DSD Registry. J Clin Endocrinol Metab. 2014;99(2):E348-55.
22. Paula GB, Barros BA, Carpini S, Tincani BJ, Mazzola TN, Guaragna MS, et al. 408 cases of genital ambiguity followed by single multidisciplinary team during 23 years: etiologic diagnosis and sex of rearing. Int J Endocrinol. 2016;496374.
23. Robboy SJ, Jaubert F. Neoplasms and pathology of sexual developmental disorders (intersex). Pathology. 2007;39:147-63.
24. Cools M, Drop SL, Wolffenbuttel KP, Oosterhuis JW, Looijenga LH. Germ cell tumors in the intersex gonad: old paths, new directions, moving frontiers.Endocr Rev. 2006;27:468-84.
25. Kuhnle U, Krahl W. The impact of culture on sex assignment and gender development in intersex patients. Perspect Biol Med. 2002;45:85-103.
26. Spinola-Castro AM. Importance of ethical and psychological features in the intersex management. Arq Bras Endocrinol Metabol. 2005;49:46-59.
27. Daaboul J, Frader J. Ethics and the management of the patient with intersex: a middle way. J Pediatr Endocrinol Metab. 2001;14:1575-83.
28. Kirtane J. Ethics in intersex disorders. Issues Med Ethics. 2000;8:47-8.
29. Hester JD. Intersex(es) and informed consent: how physicians' rhetoric constrains choice. Theor Med Bioeth. 2004;25:21-49.
30. Hermer L. Paradigms revised: intersex children, bioethics & the law. Ann Health Law. 2002;11:195-236.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()