Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 19(3) - Setembro 2019
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Sindrome DOORS: Diagnóstico Diferencial nas Sindromes Epilépticas com Displasia Ungueal
DOORS syndrome: Differential Diagnosis in Epileptic Syndromes with Nail Dysplasia
Geovanna Mazza Morais1; Sofia Abrão Lucante Gonçalves1; Marcela Almeida1; Laura Vagnini2; Zumira Aparecida Carneiro1; Regina Albuquerque3; Charles Marques
DOI:10.31365/issn.2595-1769.v19i3p69-73
1. Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
2. Centro Paulista de Diagnostico e Pesquisa, Genetica Clínica - Ribeirão Preto - SP - Brasil
3. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
Endereço para correspondência:
Recebido em: 23/06/2019
Aprovado em: 05/03/2020
Instituição: Centro Universtario Estacio de Ribeirao Preto, Faculdade de Medicina - Ribeirão Preto - SP - Brasil
Resumo
INTRODUÇÃO: A síndrome DOORS é uma rara doença genética, acometendo menos de um paciente em cada 1.000.000, consistindo em uma enfermidade multissistêmica cujas primeiras manifestações clínicas costumam estar presentes ao nascimento e podem ser facilmente identificadas ao exame físico do neonato.
OBJETIVO: Relatar caso de paciente brasileira com características clínicas compatíveis com síndrome DOORS cujo exame genético-molecular não evidenciou mutações no gene TBC1D24, ampliando a heterogeneidade genética nessa doença.
DESCRIÇÃO DO CASO: Relatamos caso de paciente do sexo feminino, 1 ano 7 meses, apresentando hipertrofia gengival, crises convulsivas, onicodistrofia, atraso do desenvolvimento, paralisia no sexto par craniano à direita encaminhada para avaliação de possível etiologia genética para seu quadro clínico. Exames de cariótipo convencional, array cromossômico e sequenciamento completo de exoma foram compatíveis com a normalidade; sequenciamento do gene TBC1D24 não apontou presença de mutações nesse gene.
DISCUSSÃO: A síndrome DOORS é um distúrbio congênito que envolve múltiplas anormalidades, entre elas: surdez, onicodistrofia, osteodistrofia, deficiência intelectual e crises convulsivas. Mutações no gene TBC1D24 podem ser encontradas em cerca de metade dos pacientes com características clínicas típicas da doença. A ausência de mutações nesse gene no presente caso sugere que mutações em outros genes podem também ser responsáveis pelo fenótipo DOORS.
Palavras-chave: Sequenciamento de Nucleotídeos em Larga Escala; Epilepsia Generalizada; Deficiências do Desenvolvimento; Crescimento Excessivo da Gengiva; Deficiência Intelectual
Abstract
INTRODUCTION: DOORS syndrome is a rare genetic disease, affecting 1 patient in 1.000.000, with multisystemic involvement whose first clinical manifestations usually are present at birth and can be easily identified in the newborn physical examination.
OBJECTIVE: To report a Brazilian patient with features of DOORS syndrome whose genetic molecular testing has not shown TBC1D24 gene mutations, widening the genetic heterogeneity in such disease.
CASE DESCRIPTION: Female case, 1 year 7 months, presenting with gingival hypertrophy, seizures, onicodystrophy, developmental delay, right sixth cranial nerve palsy referred for genetic investigation. Conventional karyotype, chromosome array and whole exome sequencing were unremarkable; TBC1D24 gene sequencing did not show abnormal findings.
DISCUSSION: "DOORS" syndrome is a congenital disorder involving multiple abnormalities: deafness; onychodystrophy; osteodystrophy; intellectual disability and seizures. TBC1D24 mutations can be found in about half of the patients with typical clinical features of the disease. Absence of mutations in this gene in our case suggests that other genes may be responsible for DOORS phenotype.
Keywords: Nucleotide Mapping; Epilepsy, Generalized; Developmental Disabilities; Gingival Hypertrophy; Intellectual Disability
INTRODUÇÃO
A síndrome DOORS é uma rara doença genética, acometendo menos de um paciente em cada 1.000.000, consistindo em uma enfermidade multissistêmica cujas primeiras manifestações clínicas costumam estar presentes ao nascimento1.
DOORS é um acrônimo englobando Deafness (surdez), Onychodystrophy (onicodistrofia), Osteodystophy (osteodistrofia), Mental Retardation e Seizures (convulsões). As manifestações clínicas principais são a combinação de surdez neurosenssorial, encurtamento das falanges distais com hipoplasia ungueal em mãos e pés, associada à deficiência intelectual e crises convulsivas2. Aproximadamente metade dos pacientes com as características clinicas da doença apresentam mutações no gene TBC1D24 e a outra metade pode ter como causa do fenótipo clinico outro gene ainda não descoberto ou ser causado por outra doença que possui características que se sobrepõem às da síndrome DOORS3.
Normalmente, pacientes desenvolvem surdez congênita bilateral do tipo neurossensorial. Além disso, pacientes com DOORS apresentam alterações na textura, cor e estrutura das unhas, que podem ficar disformes, descoradas e, principalmente, hipoplásicas4. Os dedos dos pés e das mãos podem apresentar deformidades ósseas: os polegares são longos, e possuem aumento anormal dos ossos da falange distal. Em alguns casos, é possível estar presente, ainda, um terceiro osso (extranumerário) que pode não estar totalmente formado ou ter alguma malformação5.
A deficiência intelectual também está presente em pacientes com a síndrome, podendo variar de grau leve a profundo e, em alguns casos, resultar em atraso na obtenção de marcos do desenvolvimento, como andar, sentar e falar6. Durante o primeiro ano de vida, alguns lactentes afetados podem apresentar episódios súbitos de atividade elétrica descontrolada no cérebro, principalmente convulsões do tipo grande mal. Pacientes com DOORS podem exibir diferentes tipos de crises convulsivas, como tônico-clônicas generalizadas, crises focais e mioclônicas de início precoce.6
Relatamos caso de paciente brasileira com características clínicas compatíveis com síndrome DOORS cujo exame genético-molecular não evidenciou mutações no gene TBC1D24, explorando o diagnóstico diferencial com outras síndromes epilépticas que possam cursar com alteração ungueal. Neste relato, os dados foram obtidos através da revisão do prontuário com a autorização do comitê de ética do hospital e do paciente, obtida por assinatura do termo de consentimento livre e esclarecido, com autorização por escrito dos genitores para utilização de imagens da paciente para a realização desse artigo.
DESCRIÇAO DO CASO
Paciente criança, sexo feminino, 1 ano e 7 meses de idade, filha de um casal jovem e não consanguíneo, foi encaminhada para avaliação por apresentar quadro de atraso neuropsicomotor associado a crises convulsivas. Trata-se de paciente nascida de parto cesáreo a termo (38 semanas e 1 dia), após uma gestação sem intercorrências. Nasceu com peso de 2770 g, comprimento 48 cm, perímetro cefálico 33 cm, perímetro torácico 31 cm, com Apgar 5 no 1º minuto e 7 no 5º minuto.
Ao nascimento, contudo, apresentou sinais de anóxia, evoluindo com parada cardiorrespiratória, levando-a direto para UTI onde ficou internada 23 dias. Na UTI, fazia apnéias (com duração em torno de 20 segundos), necessitando de monitorização contínua.
Apresentou broncoaspiração durante a internação, além de infecção no cordão umbilical, sendo necessário uso de antibióticos (cefepime e oxaciclina) intravenoso por 15 dias; após esse período, teve alta do hospital. Ausência de unhas nas mãos e nos pés já foi observada durante essa internação.
Por volta dos 2 meses e meio, paciente foi encaminhada para avaliação médica com história de episódios de convulsões, sem qualquer doença precipitante concomitante. Foi internada pela médica neurologista, apresentando três crises convulsivas em 10 minutos, anemia e desidratação.
Na sua evolução clínica, foi diagnosticada com laringotraqueomalácia o que levou à pneumonia aspirativa e colocação de sonda nasoentérica, usada até os 5 meses de idade; posteriormente, corrigiu-se cirurgicamente a laringotraqueomalácia, proporcionando alimentar-se novamente por via oral.
Não havia histórico familiar de qualquer deficiência intelectual, convulsões, defeitos congênitos ou quaisquer outros distúrbios genéticos. Exame citogenético (cariótipo) realizado à época foi compatível com a normalidade para o sexo feminino.
Ao exame físico, apresenta normocefalia, hipotonia global, presença de sialorreia, sem dismorfias faciais mais relevantes, presença de hipertrofia gengival (Figura 1a), discreta úvula bífida, 4 graus de hipermetropia, paralisia do sexto par no olho direito, frouxidão ligamentar, hipoplasia ungueal em dedos das mãos e agenesia de unhas em dedos dos pés (Figuras 1b e 1c).
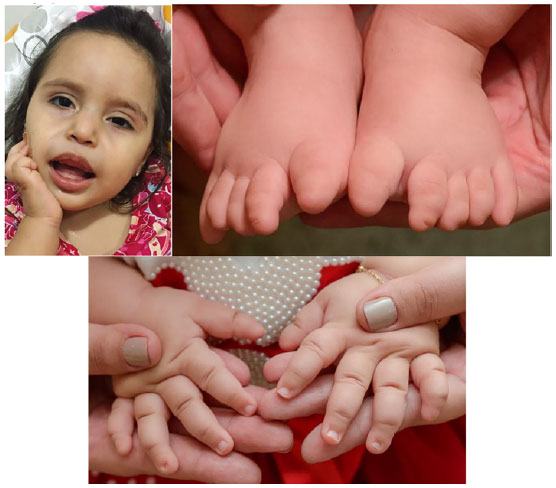
Figura 1: Presença de hipertrofia gengival na paciente e assimetria ocular pela paralisia do sexto par à direita (1a) Ausência de unhas nos dedos dos pés (1b) e hipoplasia ungueal em algumas unhas de quirodáctilos (1c). Fonte: Os Autores (2019).
Em tratamento com fisioterapia, fonoaudiologia, dieta sem lactose, uso de óculos, e em uso dos medicamentos Levetiracetam (150mg 12/12h), Valproato de sódio (125mg 12/12h), Vitamina C(20 gotas 1 vez ao dia), Furoato de Fluticasona (1 puff em cada narina antes de dormir), Propionato de Fluticasona(50 mcg 2 puffs 12/12h) e Polietilenoglicol (6 gramas na primeira mamadeira).
Exames radiográficos subsequentes evidenciaram: ausência de anormalidades em tórax e crânio; distensão gasosa com estase fecal em seguimentos do cólon; presença de osteopenia em membros superiores e inferiores; ausência de falanges distais no segundo, terceiro, quarto e quinto dedos e hipoplasia da falange distal do primeiro dedo nos dedos dos pés esquerdo e direito (Figura 2A); ausência de falange distal do quinto dedo e hipoplasia de falange distal no quarto dedo em ambas as mãos. (Figura 2B).
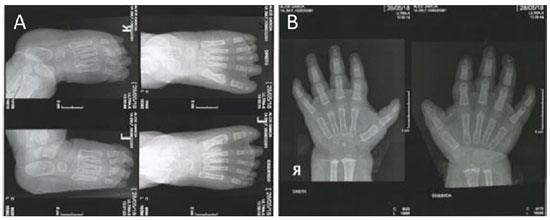
Figura 2: (A) Radiografia dos pés direito e esquerdo evidenciando ausência de falanges distais no segundo, terceiro, quarto e quinto dedos e hipoplasia da falange distal do primeiro dedo. (B) Radiografia dos dedos das mãos direita e esquerda evidenciando ausência de falange distal do quinto dedo e hipoplasia de falange distal no quarto dedo. Fonte: Os Autores (2019).
Exames de ultrassonografia abdominal e de rins/vias urinárias, ecocardiograma e tomografia de ouvidos não revelaram alterações. Perda auditiva neurossensorial bilateral leve à moderada foi evidenciada em exame de potenciais auditivos evocados (BERA).
Exame genético-molecular por sequenciamento de nova geração para o gene TBC1D24 não evidenciou variantes patogênicas no gene, sendo indicado estudos complementares com técnicas de SNP-array cromossômico e sequenciamento completo de exoma para elucidação diagnóstica com resultados normais, com boa cobertura da região de interesse e exclusão de microduplicações e microdeleções.
DISCUSSÃO
A síndrome DOORS é uma condição genética extremamente rara com menos de 50 casos relatados até a data da sua primeira descrição em 1961.5 Cantwell (1975) descreveu a recorrência dessa incomum associação de surdez, onicodistrofia, osteodistrofia e deficiência intelectual em um outro paciente.5
As anormalidades características associadas à síndrome são: surdez, devido a um defeito do ouvido interno ou nervo auditivo (perda auditiva neurossensorial); onicodistrofia ou malformação das unhas; osteodistrofia (malformação de ossos) e deficiência intelectual.8 Crises convulsivas são comumente descritas em pacientes com síndrome DOORS, mas não sao observados em todos os pacientes.8
Em estudo recente, descobriu-se que mutações no gene TBC1D24 estão presentes em uma grande parte dos pacientes com o fenótipo DOORS, mas ainda há provavelmente outros genes responsáveis por essa doença que ainda não foram identificados.5 O gene TBC1D24 codifica proteínas ativadoras de GTPases que coordenam o transporte das vesículas intracelulares. Também exerce papel na maturação morfológica e funcional do circuito neuronal, o que pode explicar os distúrbios epilépticos. Observou-se , em modelos animais, a expressão do gene TBC1D24 foi encontrada em condrócitos das falanges distais do membro anterior do camundongo.8
A surdez neurossensorial, um dos elementos mais característicos dessa síndrome, é profunda e bilateral, podendo ser considerada praticamente universal na síndrome DOORS , sendo usualmente congênita. No entanto, existe variabilidade na apresentação dessa perda auditiva.8
Razões para a coexistência de unhas, ossos e anormalidades auditivas pode ser encontradas com base embriológica.8 A formação óssea ocorre de duas maneiras: ossificação intramembranosa e ossificação endocondral. A maioria dos ossos chatos, como os ossos do crânio, formam-se diretamente das células mesenquimais por ossificação intramembranosa.8 No entanto, no labirinto há uma exceção, e sua embriologia é semelhante à da coluna vertebral, costelas e membros. A cartilagem finalmente é substituída por osso, o que é chamado de ossificação endocondral. Assim, a osteodistrofia e anormalidades no labirinto podem coexistir, porque ambos se originam do mesoderma e são formados pela ossificação endocondral.8
A onicodistrofia, nessa doença, manifesta-se sob a forma de anoníquia, hipoplasia, unhas rudimentares, descoloridas com estrutura anormal na forma e textura em uma ou todas as unhas.5 Osteodistrofia pode ser observada sob a forma de falanges distais ausentes (como no caso da paciente do relato) ou displásicas.5
As convulsões na síndrome DOORS são uma manifestação clínica proeminente, são descritas em 88% dos casos e podem ser de natureza progressiva e gravidade crescente, embora nao haja um padrão específico de tipo de crise (como do tipo mioclônica visto em outras doenças genéticas), as mesmas são descritas usualmente como generalizadas.8 A deficiência intelectual pode ser leve à profunda, freqüentemente associada ou decorrente das crises convulsivas.7 Anormalidades oftalmológicas, como atrofia óptica, podem ser observadas em alguns pacientes7. Na paciente do relato, não se identificaram lesões do nervo óptico, porém ela possui estrabismo decorrente da paralisia do sexto par do olho direito. Trata-se de um achado ainda não previamente descrito na síndrome DOORS, o que reforça a variabilidade de expressão clínica nessa enfermidade e a expansão de mais um aspecto fenotípico dessa síndrome a partir desse relato de caso.
No diagnóstico diferencial da síndrome DOORS, estariam outras doenças com sobreposiçao clínica por apresentarem displasia ungueal associada a atraso de neurodesenvolvimento e crises convulsivas, como a síndrome de Coffin-Siris, a síndrome de Temple-Baraitser e a síndrome de Carpenter tipo 2, entre outras.3
No caso relatado, a paciente apresenta as principais manifestações da sindrome incluindo onicodistrofia, osteodistrofia e deficiência intelectual. A paciente também apresentava crises convulsivas graves, manifestação presente na maioria dos casos de DOORS. No entanto, a mutação do gene TBC1D24 característica da doença não foi observada na investigação inicial da paciente, corroborando a hipótese de que outros genes podem estar envolvidos na síndrome.
Em relação ao mecanismo de herança da síndrome DOORS, considera-se tratar-se de enfermidade com herança autossômica recessiva, sendo fundamental o aconselhamento genético para os genitores, visto que, nesse tipo de herança, o risco empírico de recorrência da doença em futura prole do casal seria de 25%, mesmo não encontrando variantes no gene principal envolvido. Infelizmente, por não terem sido identificadas variantes no gene TBC1D24 , não há possibilidade de diagnóstico pré-natal ou pré-implantação para o casal.
Embora rara, sinais e sintomas da síndrome DOORS permitem o diagnóstico precoce e encaminhamento oportuno para a equipe médica multidisciplinar que deve ser envolvida no manejo desses pacientes, como a otorrinolaringologia, neurologia pediátrica, genética médica, fonoaudiologia e fisioterapia, entre outros.
REFERÊNCIAS
1.Campeau,P. DOOR syndrome, 2019 https://www.orpha.net/consor/cgi-bin/OC_Exp.php?lng=EN&Expert=79500
2. Michalek P. Anaesthetic management of an adult patient with DOOR syndrome: a case report, 2009. https://www.ncbi.nlm.nih.gov/pubmed/19830001
3. Campeau P.M. The genetic basis of DOORS syndrome: an exome-sequencing study,2013. https://www.ncbi.nlm.nih.gov/pubmed/24291220
4. Aaron W.J. DOOR syndrome: Clinical Report, Literature Review and Discussion of Natural History, 2007. https://www.ncbi.nlm.nih.gov/pubmed/17994565
5. Lal. D.N. Absence of Nails, Deaf Mutism, Seizures, and Intellectual Disability: a case report, 2015. https://www.ncbi.nlm.nih.gov/pubmed/26023614
6. Lozano R. Clinical Intrafamilial Variability in Lethal Familial Seizure Disorder caused by TBC1D24 Mutations, 2015. https://www.ncbi.nlm.nih.gov/pubmed/27541164
7. Devayanivasudevan L. Absense of nails, deaf-mutism, seizures, and intellectual disability: a case report, 2015. https://www.ncbi.nlm.nih.gov/pubmed/26023614
8. Santos M. DOOR syndrome: A case report and its embryological basis, 2019. https://www.ncbi.nlm.nih.gov/pubmed/30579089
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()