Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 18(1) - Março 2018
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
RELATOS DE CASO
Neurofibromatose tipo 2: relato de caso na infância
Neurofibromatosis type 2: case report in childhood
Danila de Souza Carraro1; Priscila Paula Pires2; Alessandra Kiesewetter3; Clarissa Bueno4; Sabrina Bortolin Nery5; Nelson Kazunobo Horigoshi6
1. Doutora em Ciências Médicas pela Faculdade de Medicina da Universidade de Sao Paulo. Pediatra e Pneumologista Infantil do Hospital Infantil Sabará
2. Radiologista do Hospital Infantil Sabará
3. Otorrinolaringologista do Hospital Infantil Sabará
4. Doutora em Ciências Médicas pela Faculdade de Medicina da Universidade de Sao Paulo. Neurologista do Hospital Infantil Sabará
5. Gastropediatra. Diretora Clínica do Hospital Infantil Sabará
6. Pediatra e Intensivista Pediátrico. Gestor da Unidade de Terapia Intensiva do Hospital Infantil Sabará
Endereço para correspondência:
Recebido em: 25/8/2017
Aprovado em: 31/10/2017
Instituição: Fundação José Luiz Egydio Setúbal/Hospital Infantil Sabará
Resumo
INTRODUÇÃO: a neurofibromatose tipo 2 é uma doença autossômica dominante, causada por mutação no gene NF2, localizado no cromossomo 22q12.2, que codifica a proteína neurofibromina-2, também denominada merlin. Clinicamente, a doença caracteriza-se por múltiplos tumores do sistema nervoso central, predominando schwannomas, meningiomas e ependimomas. Raros casos são descritos com apresentação no primeiro ano de vida.
OBJETIVO: descrever o caso de uma paciente na faixa etária pediátrica no qual a anamnese e o exame físico detalhado contribuíram para o diagnóstico precoce de neurofibromatose tipo 2.
DESCRIÇÃO DO CASO: paciente de 7 anos, sexo feminino, internada com diagnóstico inicial de síndrome vestibular secundária e otite média aguda. Durante a internação e evolução clínica, notou-se o surgimento de nistagmo horizonto-vertical e anisocoria com midríase, levando a maior investigação do caso e ao diagnóstico precoce de neurofibromatose tipo 2.
DISCUSSÃO: neurofibromatose tipo 2, doença caracterizada por múltiplos tumores do sistema nervoso central, também pode apresentar alterações oculares, incluindo catarata de início precoce, tumores da bainha do nervo óptico, hamartomas e membranas retinianas. Até o momento, ainda há restrições para estabelecer os critérios clínicos quanto à sensibilidade para diagnosticar todos os indivíduos de fato acometidos pela doença. Esta consideração é particularmente importante nas crianças, que podem ainda não ter evidentes todas as manifestações clínicas que permitiriam o diagnóstico, sendo necessário seguimento clínico e de imagem e, eventualmente, diagnóstico evolutivo.
Palavras-chave: Neoplasias do sistema nervoso central; Neurofibromatose 2; Criança.
Abstract
INTRODUCTION: neurofibromatosis type 2 is an autosomal dominant disease, caused by a mutation in the NF2 gene located on chromosome 22q12.2, which encodes the neurofibromin-2 protein, also called merlin. Clinically, the disease is characterized by multiple tumors of the central nervous system, predominating schwannomas, meningiomas and ependymomas. Cases of this disease in children are rarely described in the first year of life.
OBJECTIVE: describe the case of a patient in the pediatric age group where the anamnesis and detailed physical examination contributed to the early diagnosis of neurofibromatosis type 2.
CASE DESCRIPTION: a 7-year-old female patient, hospitalized with an initial diagnosis of vestibular syndrome secondary to acute otitis media. During hospitalization and clinical evolution, the appearance of horizontal-vertical nystagmus and anisocoria with mydriasis led to further investigation of the case and the early diagnosis of neurofibromatosis type 2.
DISCUSSION: neurofibromatosis type 2 disease, characterized by tumors of the central nervous system, may also present eye lesions, including an early onset cataract, optic nerve sheath tumors, hamartomas and retinal membranes. To this date, there are still restrictions to establish the clinical criteria regarding the sensitivity to diagnose all the individuals actually affected by the disease. This consideration is particularly important in children, who may not yet have all the clinical manifestations that would allow the diagnosis, requiring clinical and imaging follow-up and, eventually, an evolutionary diagnosis.
Keywords: Central nervous system neoplasms; Neurofibromatosis 2; Child.
INTRODUÇÃO
A neurofibromatose tipo 2 (NF2) é uma doença autossômica dominante, com incidência estimada de 1:40.000 nascidos,1 causada por mutação no gene NF2, localizado no cromossomo 22q12.2, que codifica a proteína neurofibromina-2, também denominada merlin. Esta faz parte de um grupo de proteínas de ligação entre o citoesqueleto e a membrana plasmática, regulando a proliferação celular por meio da ação em diversas cascatas proteicas.2
Clinicamente, a doença caracteriza-se por múltiplos tumores do sistema nervoso central, predominando schwannomas, meningiomas e ependimomas.3 Alterações oculares também são comuns, incluindo catarata de início precoce, tumores da bainha do nervo óptico, hamartomas e membranas retinianas.4 A maioria dos casos tem acometimento difuso, mas alguns podem apresentar tumores unilaterais ou lesões restritas a regiões delimitadas, caracterizando as formas em mosaico ou segmentares da NF24. Embora se manifeste mais frequentemente na vida adulta, cerca de 18% dos casos apresentam-se antes dos 15 anos,5 e raros são descritos com apresentação no primeiro ano de vida.6
O presente estudo tem como objetivo discutir o caso de uma paciente na faixa etária pediátrica com neurofibromatose tipo 2.
Este trabalho foi aprovado pelo Comitê de Ética em Pesquisa da Comissão Nacional de Ética em Pesquisa (Conep), sob o Certificado de Apresentação para Apreciação Ética (CAAE): 67951417.8.0000.5567.
DESCRIÇÃO DO CASO
Paciente de 7 anos, sexo feminino, caucasiana, que apresentava febre há seis dias e perda do equilíbrio. No exame físico, notou-se hiperemia e abaulamento de membrana timpânica com secreção retrotimpânica bilateral, mais evidente à esquerda, além de nistagmo horizontal para o lado direito, feito diagnóstico de otite média aguda e síndrome vestibular secundária, internada para antibioticoterapia endovenosa com ceftriaxone, corticoterapia e timpanotomia. Tomografia computadorizada de mastoide realizada no primeiro dia de internação descartou mastoidite aguda e a paciente evoluiu afebril após 24 horas, com remissão da ataxia após a timpanotomia. Entretanto, devido ao surgimento de nistagmo horizonto-vertical, anisocoria com midríase e ausência de reflexo fotomotor à direita, foi solicitada avaliação da equipe de neurologia pediátrica.
O exame físico neurológico evidenciava força muscular globalmente preservada, reflexos osteotendíneos normoativos, equilíbrio e coordenação sem alterações. Ao exame dos nervos cranianos, observamos paresia do III nervo à esquerda, caracterizada por estrabismo divergente e paresia de III nervo à direita, caracterizada por deficit do olhar para cima, semiptose, anisocoria, com midríase à direita e reflexo fotomotor lentificado. O teste de Weber com diapasão indicava perda condutiva do lado esquerdo e fundoscopia com suspeita de lesão retiniana de origem tumoral à esquerda.
A paciente tinha diagnóstico prévio de coriorretinite em olho esquerdo decorrente de toxoplasmose congênita, diagnosticada no primeiro ano de vida, apresentando desde então o estrabismo divergente observado ao exame neurológico. Após 24 horas do término do tratamento medicamentoso, administrado durante dez dias, a paciente evoluiu com paralisia facial periférica súbita à esquerda grau IV pela Escala de House-Brackmann, sendo reintroduzido o corticoide e mantido por 21 dias.
Foram solicitadas tomografia computadorizada (TC) de crânio e mastoide com contraste, que evidenciaram otomastoidopatia inflamatória bilateral, alargamento do gânglio geniculado do nervo facial esquerdo, pansinusopatia inflamatória aguda, calcificações alongadas no cerebelo, bem como lesões calcificadas na topografia do cavo de Meckel esquerdo que alargava o respectivo forame oval e na porção superior da cisterna pontocerebelar deste lado. As calcificações cerebelares eram inespecíficas, assim como as alterações orbitais.
Posteriormente foi submetida à ressonância magnética (RM) de crânio, mastoides e órbitas, que demonstrou proeminências das câmaras anteriores das órbitas, aparente espessamento/realce das paredes posteriores dos globos oculares, sobretudo à esquerda, nervos ópticos discretamente assimétricos, observando-se à esquerda menor espessura e maior sinal em T2, lesões com realce nos condutos auditivos internos (CAI), maior à esquerda; lesões na topografia do cavo de Meckel esquerdo que se estende para o forame oval e junto aos trajetos cisternais dos nervos oculomotores, além de lesão no pedúnculo cerebelar médio direito e lesão com realce na foice cerebral.
Na RM de coluna total identificou-se uma pequena lesão em D3-D4 intradural extramedular e outra lesão foraminal esquerda D5-D6, ambas com realce pelo contraste. A análise conjunta dos exames de imagens sugeriu que as lesões nos CAI e foraminal na coluna dorsal, bem como o alargamento do gânglio geniculado do facial esquerdo são compatíveis com schwannomas, enquanto aquelas no cavum de Meckel, nos trajetos cisternais do oculomotor, na foice cerebral e a lesão extramedular intradual são mais compatíveis com meningiomas (Figura 1).
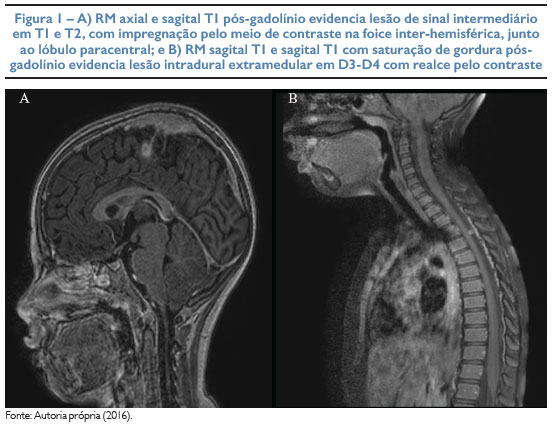
A lesão hiperintensa no pedúnculo cerebelar médio direito tem características que podem ser encontradas em hamartoma/vacuolização de mielina e em neoplasia de origem glial.
Após exames de imagem e associação com critérios clínicos, confirmou-se o diagnóstico de NF2.
DISCUSSÃO
Relatamos o caso de uma paciente com um quadro de otite média aguda e sintomas compatíveis com labirintite tóxica/infecciosa, no qual o exame da motricidade ocular revelou novas alterações que levaram à investigação detalhada através de neuroimagem, permitindo o diagnóstico incidental de NF2.
A avaliação inicial na suspeita de NF2 inclui exames para confirmar o diagnóstico e excluir complicações.4 Devem ser realizados anamnese detalhada, pesquisa de lesões cutâneas, exame neurológico, oftalmológico e investigação específica da função auditiva e vestibular. A ressonância de crânio e coluna cervical deve ser realizada para identificação dos schwannomas, particularmente do nervo vestíbulo-coclear, mas também em outras regiões do tronco encefálico, bem como identificação de outros possíveis tumores associados a esta condição, como meningiomas e ependimomas, que ocorrem predominantemente na região paravertebral. A ressonância da coluna torácica e lombar é indicada de rotina em alguns serviços, mas pode ser reservada nos casos com alterações sugestivas no exame clínico. O exame oftalmológico deve incluir o fundo de olho para pesquisa de catarata, presente em 40% dos pacientes, hamartomas de retina e membranas epirretinianas. A investigação audiológica deve ser feita pela audiometria tonal e vocal, podendo ser completada com potenciais evocados auditivos de tronco encefálico.4,7
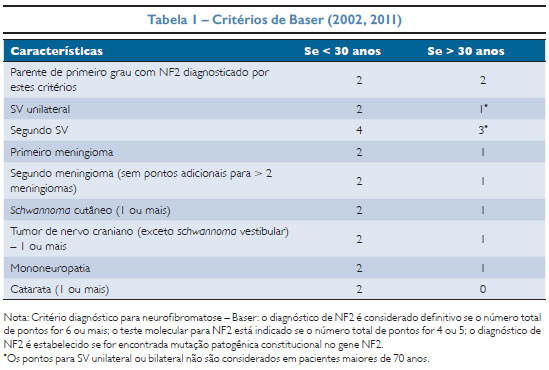
O diagnóstico da NF2 é baseado em critérios clínicos, definidos pelo National Institute of Health (NIH), revisados em 1990,8 e os critérios de Manchester.9 Os critérios estabelecidos até o momento têm restrições quanto à sensibilidade para diagnosticar todos os indivíduos de fato acometidos pela NF2. Esta consideração é particularmente importante nas crianças, que podem ainda não ter evidentes todas as manifestações clínicas que permitiriam o diagnóstico, sendo necessário seguimento clínico e de imagem e, eventualmente, diagnóstico evolutivo. Diante desta limitação, Baser e colaboradores10 propuseram um conjunto de critérios diagnósticos (Tabela 1), levando em consideração a história natural e aspectos genéticos da NF2, procurando melhorar a sensibilidade diagnóstica, particularmente para a população pediátrica. A nossa paciente preenche a condição A para o diagnóstico de NF2 segundo os critérios de Manchester e atinge pontuação 10 segundo os critérios de Baser.
O estudo molecular também pode confirmar o diagnóstico pela identificação de mutação patogênica no gene NF2. Entretanto, o diagnóstico molecular não substitui os critérios clínicos, de modo que a mutação pode ser detectada apenas no tecido tumoral e um exame negativo em sangue periférico não exclui a condição.6,11
Entre os diagnósticos diferenciais a ser considerados na presença de múltiplos schwannomas, temos a schwannomatose, caracterizada por múltiplos schwannomas de nervos periféricos, sem envolvimento concomitante do nervo vestibular.11 Nesta condição os schwannomas intracranianos ocorrem em 10% dos casos12 e lesões retinianas não são descritas. Os critérios diagnósticos incluem: 1) pelo menos dois schwannomas não dermais confirmados por biópsia, sem evidência radiológica de schwannoma vestibular (SV) bilateral; ou 2) um schwannoma não dermal ou meningioma intracraniano e um parente de primeiro grau com schwannomatose.13 O diagnóstico da schwannomatose também pode ser feito por teste molecular, pela identificação de mutação patogênica nos genes SMARCB1 ou LZTR1.12,13
Embora seja uma condição caracteristicamente de apresentação na vida adulta, cerca de 18% dos indivíduos com NF2 têm manifestação clínica antes dos 15 anos. As formas de manifestação dividem-se em três grupos, sendo dois de início na faixa etária pediátrica:
a) leve ou tipo Gardner – de início na vida adulta, frequentemente com VS bilateral como única manifestação;
b) grave ou tipo Wishart – de início na infância e adolescência, com múltiplos tumores de sistema nervoso central, rapidamente progressivos e, geralmente, tendo como primeira manifestação tumores de outros tipos histológicos, diferentes do schwannoma;
c) NF2 congênita – de início nos primeiros meses de vida, podendo apresentar como primeira manifestação clínica lesões oculares ou cutâneas e o VS como achado incidental.4,5
Há poucos casos relatados de NF2 congênita, sendo a casuística e revisão de Ruggieri e colaboradores,6 com descrição de três casos e levantamento de outros três pacientes da literatura, a principal fonte de informação. A forma de apresentação e história natural da NF2 congênita é variável e distingue-se das formas de apresentação em outras faixas etárias pediátricas. Os sintomas iniciais e que conduzem à investigação são habitualmente alterações da retina e/ou opacidade do cristalino e lesões cutâneas.
As lesões cutâneas da NF2 na infância apresentam-se como placas múltiplas em localizações atípicas (mãos, pernas, joelhos e face) e manchas café com leite. Estas lesões tendem a regredir e desaparecer, paralelamente a uma progressão do SV. O SV pode ser identificado nos exames de imagem tão precocemente quanto aos 4 meses de idade, podendo se manter estável e assintomático por longo período, sendo característica a sua progressão a partir da puberdade, associando-se, a partir de então, com sintomas clínicos, particularmente a perda auditiva.
Acreditamos que a nossa paciente seja um dos raros casos de NF2 congênita, com a lesão retiniana inicialmente diagnosticada de modo equivocado como coriorretinite por toxoplasmose.
O seguimento clínico na NF2 deve ser realizado com reavaliações a cada três a seis meses, com exame neurológico completo. A ressonância de crânio e coluna deve ser realizada periodicamente, bem como o exame audiológico e o exame oftalmológico.3,4
Devido à multiplicidade de tumores cranianos, espinhais e em nervos periféricos, a remoção de todas as lesões não é possível, nem mesmo indicada. O objetivo primário deve ser preservar a função e melhorar a qualidade de vida. A cirurgia ainda é o tratamento de escolha para os schwannomas de VIII par e para os outros tumores de SNC4.
No caso dos schwannomas de VIII par, consensos mais recentes propõem a ressecção dos tumores maiores que 3 cm, permitindo a preservação da função do nervo facial e proteção do tronco encefálico.5 O objetivo da cirurgia pré-sintomática é preservar a audição e minimizar outras complicações, como paralisia facial e disfagia. O acesso suboccipital para pequenos tumores pode preservar a audição. Tumores multilobulados ou maiores que 1,5 cm podem ser seguidos e ressecados quando houver outra indicação para cirurgia, como aumento de pressão intracraniana ou sintomas neurológicos, devido ao risco de sequelas pós-operatórias. Schwannomas de outros pares cranianos geralmente têm crescimento lento e são oligossintomáticos, sendo indicada ressecção no caso de sintomas neurológicos importantes ou crescimento rápido.4
Tumores medulares, como os ependimomas, geralmente respondem bem à cirurgia, sem necessidade de abordagens repetidas ou recidiva.5 Em contrapartida, tumores pontinos e bulbares (geralmente ependimomas e astrocitomas) frequentemente necessitam de ressecções parciais repetidas com sequelas severas.
Irradiação estereotáxica pode ser usada como alternativa ou associada à cirurgia, no entanto sabe-se que pode aumentar o risco de malignidade secundária. A radioterapia para schwannoma vestibular na NF2 tem piores resultados em comparação com tumores esporádicos.4,7
Até o momento, não há quimioterapia comprovadamente efetiva para o tratamento dos tumores relacionados à NF2.7
Recentemente, o uso de terapias biologicamente direcionadas utilizando moléculas foi impulsionado pelo maior conhecimento das vias moleculares envolvidas na patogênese da NF2. Foram empregadas com sucesso, porém devido ao uso muito recente e em limitado número de pacientes, ainda estão sob estudo.7
A sobrevida média após o diagnóstico é de cinco anos em 85%, dez anos em 67% e 20 anos em 38% dos pacientes.14 Menor idade ao diagnóstico e presença de meningiomas intracranianos são associados a aumento de mortalidade.15
No caso dos schwannomas vestibulares, a taxa de crescimento do tumor foi inversamente relacionada com a idade de início dos sintomas e diretamente relacionada ao volume inicial do tumor. A ressecção cirúrgica pode acelerar a taxa de crescimento de tumores contralaterais, por motivo ainda não elucidado.15
REFERÊNCIAS
1 Evans DG, Moran A, King A, Saeed S, Gurusinghe N, Ramsden R. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-7.
2 Curto M, McClatchey A. NF2/Merlin: a coordinator of receptor signalling and intercellular contact. Brit J Cancer. 2008;98(2):256-62.
3 Ardern-Holmes S, Fisher G, North K. Neurofibromatosis Type 2: presentation, major complications, and management, with a focus on the pediatric age group. Journal Child Neurol. 2017;32(1):9-22.
4 Ruggieri M, Praticò AD, Evans DG. Diagnosis, management, and new therapeutic options in childhood neurofibromatosis type 2 and related forms. Semin Pediatr Neurol. 2015;22(4):240-58.
5 Evans DGR, Birch JM, Ramsden RT. Paediatric presentation of type 2 neurofibromatosis. Arch Dis Child. 1999;81(6):496-9.
6 Ruggieri M, Gabriele AL, Polizzi A, Salpietro V, Nicita F, Pavone P et al. Natural history of neurofibromatosis type 2 with onset before the age of 1 year. Neurogenetics. 2013;14(2):89-98.
7 Ruggieri M, Pratico AD, Serra A, Mailino L, Cocuzza S, Di Mauro P et al. Childhood neurofibromatosis type 2 (NF2) and related disorders: from bench to bedside and biologically targeted therapies. Acta Otorhinolaryngol Ital. 2016;36(5):345-67.
8 Mulvihill JJ, Parry DM, Sherman JL, Pikus A, Kaiser-Kupfer MI, Eldridge R. NIH conference. Neurofibromatosis 1 (Recklinghausen disease) and neurofibromatosis 2 (bilateral acoustic neurofibromatosis). An update. Ann Intern Med. 1990;113(1):39-52.
9 Evans DG, Huson SM, Donnai D, Neary W, Blair V, Newton V et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-18.
10 Baser ME, Friedman JM, Wallace AJ, Ramsden RT, Joe H, Evans DG. Evaluation of clinical diagnostic criteria for neurofibromatosis 2. Neurology. 2002;59(11):1759-65.
11 Kresak JL, Walsh M. Neurofibromatosis: a review of NF1, NF2 and Schwannomatosis. J Pediatr Genet. 2016;5(2):98-104.
12 Kehrer-Sawatzki H, Farschtschi S, Mautner VF, Cooper DN. The molecular pathogenesis of schwannomatosis, a paradigm for the co-involvement of multiple tumour suppressor genes in tumorigenesis. Hum Genet. 2017;136(2):129-48.
13 Baser ME, Friedman JM, Evans DG. Increasing the specificity of diagnostic criteria for schwannomatosis. Neurology. 2006;66(5):730-2.
14 Otsuka G, Saito K, Nagatani T, Yoshida J. Age at symptom onset and long-term survival in patients with neurofibromatosis type 2. J Neurosurg. 2003;99(3):480-3.
15 Hexter A, Jones A, Joe H, Heap L, Smith MJ, Wallace AJ et al. Clinical and molecular predictors of mortality in neurofibromatosis 2: a UK national analysis of 1192 patients. J Med Gen. 2015;52(10):699-705.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()