Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 18(1) - Março 2018
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
RELATOS DE CASO
Relato de caso: púrpura de Henoch-Schönlein
Case report: Henoch-Schönlein purpura
Simone de Abreu Neves Salles1; Juliana Aparecida Lemos da Silva2; Larissa Verônica Kamarowski3; Thaís de Albuquerque4
1. Médica-Chefe da Unidade de Dermatologia da Universidade Federal Fluminense
2. Acadêmica de Medicina da Universidade Federal Fluminense
3. Acadêmica de Medicina da Universidade Federal Fluminense
4. Acadêmica de Medicina da Universidade Federal Fluminense
Endereço para correspondência:
Recebido em: 17/1/2018
Aprovado em: 15/2/2018
Instituiçao: Universidade Federal Fluminense
Resumo
INTRODUÇÃO: a púrpura de Henoch-Schönlein, vasculite mais comum na infância, é mediada por depósitos de imunocomplexos de imunoglobulina A, podendo ser precedida por infecções respiratórias superiores.
OBJETIVO: relatar caso com distribuição cutânea atípica pelo acometimento da face e membros superiores e ausência de lesões nas nádegas que, junto aos membros inferiores, é o local em que as lesões purpúricas são mais comumente observadas.
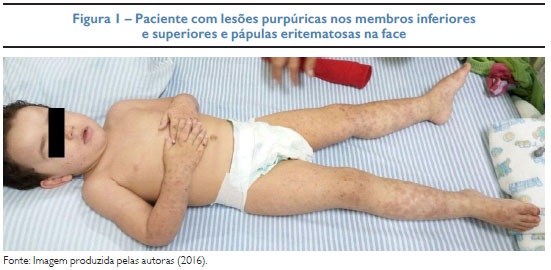
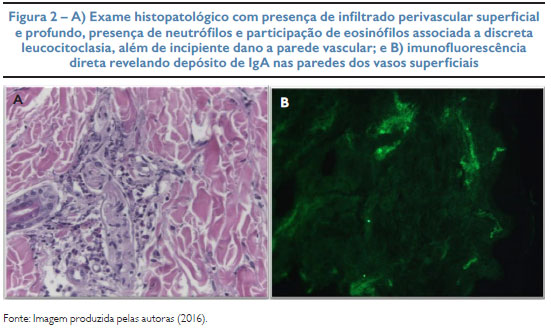
DESCRIÇÃO DO CASO: M. D. X., 2 anos e 2 meses, masculino, história prévia recente de infecção do trato respiratório superior. Paciente apresenta dor abdominal, edema e lesões purpúricas bilaterais nos membros inferiores e superiores, além de presença de pápulas eritematosas na face. Ausência de lesões cutâneas nas nádegas e tronco. Laboratorialmente, presença de leucocitose e trombocitose. O exame histopatológico revelou infiltrado perivascular superficial e profundo com presença de eosinófilos, neutrófilos e leucocitoclasia com incipiente dano à parede vascular. A imunofluorescência direta identificou depósitos de imunoglobulina A nas paredes dos vasos superficiais. Foi internado, sendo necessário apenas tratamento sintomático. Apresentou evolução satisfatória do quadro, recebendo alta hospitalar para acompanhamento ambulatorial.
DISCUSSÃO: na púrpura de Henoch-Schönlein a manifestação clínica mais frequente é a púrpura palpável atrombocitopênica, que se localiza tipicamente nas nádegas e membros inferiores. É uma doença multissistêmica, podendo afetar, além da pele, outros órgãos, como intestino, articulações e principalmente rins. O curso, em geral, é benigno, autolimitado e monocíclico, necessitando apenas de tratamento de suporte clínico.
Palavras-chave: Púrpura de Schoenlein-Henoch; Vasculite; Criança; Infecçoes respiratórias.
Abstract
INTRODUCTION: Schönlein-Henoch purpura is the most common vasculitis in children, mediated by immunoglobulin A immune complex storages, which may be preceded by upper respiratory infections.
OBJECTIVE: to report a case of an atypical cutaneous manifestation, with lesions on the face and upper limbs and absence of lesions on the buttocks, where they are commonly observed.
CASE DESCRIPTION: MDX, 2 years and 2 months old, male, recent previous history of upper respiratory tract infection. The patient presents abdominal pain, swelling and bilateral palpable purpura located on the lower and upper limbs, as well as erythematous papules on the face. Absence of cutaneous lesions on the buttocks and trunk. Laboratory exams show leukocytosis and thrombocytosis. Histopathologic exam revealed superficial and deep perivascular infiltration with eosinophils, neutrophils and leukocytoclastic vasculitis. Direct immunofluorescence study detected immunoglobulin A deposition within the walls of small vessels. He was admitted to the hospital, receiving only symptomatic treatment. The patient's condition improved and he was discharged and scheduled for outpatient follow-up.
DISCUSSION: the most frequent symptom of Schönlein-Henoch purpura is non-thrombocytopenic palpable purpura that tends to appear in areas such as buttocks and lower limbs. It is a multisystem disease, whitch affects, besides the skin, mainly intestines, joints and kidneys. This disorder often presents a benign, self-limited and monocyclic course, requiring only symptomatic treatment.
Keywords: Purpura, Schoenlein-Henoch; Vasculitis; Child; Respiratory tract infections.
INTRODUÇÃO
A púrpura de Henoch-Schönlein (PSH), ou púrpura anafilactoide, é a vasculite sistêmica mais comum na infância. Apresenta ligeira predominância no sexo masculino, curso benigno, monocíclico e autolimitado. Em 20% dos casos, podem ser observadas recorrências e/ou cronicidade.1
É uma vasculite mediada por depósito de imunocomplexos, contendo imunoglobulina A na parede de pequenos vasos. Usualmente, é precedida por uma infecção do trato respiratório superior, sendo sugerida essa condição como um fator desencadeante do quadro.2
A PHS é caracterizada pela identificação de lesão cutânea purpúrica palpável, principalmente nas nádegas e membros inferiores, podendo ser acompanhada de artralgia ou artrite, dor abdominal, sangramento intestinal e nefrite.3 Raramente, a doença pode afetar outros órgãos, como testículos, pulmões e cérebro.4
Relata-se a seguir um caso de paciente pré-escolar de 2 anos e 2 meses apresentando pápulas eritematosas na face e pápulas purpúricas nos antebraços e membros inferiores, poupando a região glútea e tronco.
DESCRIÇÃO DO CASO
Paciente de 2 anos e 2 meses, masculino, com queixa de surgimento de lesões cutâneas associadas a inapetência, vômitos e precedidas por infecção do trato respiratório superior. Na internação, apresentava irritabilidade, dor abdominal difusa, edema nas extremidades, dor à mobilização dos membros, lesões purpúricas nos membros inferiores e superiores e pápulas eritematosas na face (Figura 1). Ausência de alterações cutâneas nas nádegas e tronco. Hemograma, ureia, creatinina e elementos anormais e sedimento (EAS) sem alterações. Exame histopatológico de lesão da face interna da coxa exibiu vasculite leucocitoclástica e eosinofilia compatível com púrpura de Henoch-Schönlein. Na imunofluorescência direta, presença de depósito de IgA nas paredes dos vasos superficiais, assim como de IgM e fibrinogênio (Figura 2). Foram iniciados hidratação venosa, ranitidina, dipirona e albendazol. Evoluiu com melhora do quadro, recebendo alta hospitalar para acompanhamento ambulatorial.
DISCUSSÃO
A PHS é a vasculite mais comum na infância. Consiste na inflamação dos pequenos vasos, acometendo pele, intestino, articulações e principalmente rins.1 Predomina nas crianças na faixa etária escolar (entre 4 e 7 anos), mas pode ocorrer de 1 a 19 anos de idade.5
A causa ainda é desconhecida, no entanto existem diversos fatores desencadeantes, como: infecções das vias aéreas superiores (precedem cerca de 50% dos casos), vacinas, medicamentos, alimentos, picadas de insetos e exposição ao frio. O diagnóstico é frequentemente clínico.6
Segundo o Colégio Americano de Reumatologia, o diagnóstico de PHS caracteriza-se pela presença de púrpura palpável (manchas elevadas na pele que não desaparecem com a digitopressão) e ausência de plaquetopenia, associada a dois ou mais dos seguintes critérios: dor abdominal; idade de início (antes dos 20 anos); e alterações histopatológicas pela presença de granulócitos nas paredes de arteríolas ou vênulas.7
Para a Sociedade Europeia de Reumatologia Pediátrica, além da púrpura palpável atrombocitopênica, é necessário, no mínimo, um dos seguintes achados: dor abdominal difusa; artrites ou artralgia; acometimento renal (hematúria ou proteinúria); ou exame histopatológico com depósito predominante de imunoglobulina A na parede de pequenos vasos. De acordo com essa escola, as manifestações articulares e renais assumem importância diagnóstica.8
O achado clínico mais frequente é a lesão cutânea de púrpura palpável na ausência de plaquetopenia (encontrada em 100% dos pacientes) com localização predominantemente nos membros inferiores e nádegas, podendo ocorrer menos frequentemente na face, no couro cabeludo, nos membros superiores e raramente no tronco. As lesões surgem em surtos com intervalos de uma semana ou mais e atividade com duração entre 3 e 12 semanas.9
O edema subcutâneo é observado predominantemente nas mãos e pés, em até 30% dos pacientes. Artralgia e/ou artrite em 60 a 84% dos casos, principalmente nas articulações dos joelhos e tornozelos, o que dificulta ou impossibilita a deambulação.8
O acometimento gastrointestinal (estômago e intestino) ocorre em torno de 50 a 85% dos casos, tendo sido a apresentação inicial em até 19% dos pacientes. Os sintomas podem ser: dor abdominal, náuseas, vômitos, sangramento intestinal (enterorragia ou melena) e, raramente, perfuração intestinal. A dor abdominal varia de cólicas leves a dor intensa simulando abdome agudo.10 Além disso, geralmente é intermitente11 – podendo ser difusa ou periumbilical – e piora após as refeições.12
A nefrite ocorre em 10 a 50% dos pacientes, em geral nos três primeiros meses da doença. Doença renal grave é observada em 1 a 4,5% de todas as crianças e adolescentes com PHS, e em 7,5% são permanentes. As alterações renais mais frequentes são leves e, via de regra, evidenciadas nos exames de urina tais como hematúria, proteinúria e podem persistir por até três meses.6
As complicações mais comuns de PHS são: angina mesentérica, perfuração intestinal, obstrução duodenal, hemorragia gastrointestinal, intussuscepção, glomerulonefrite, cistite hemorrágica, síndrome nefrótica, falência renal e obstrução ureteral.9
As afecções que cursam com lesões cutâneas petequiais e purpúricas são importantes diagnósticos diferenciais. Dentre elas, podemos citar febre maculosa, endocardite bacteriana e meningococcemia. A presença de púrpura sem sintomas sistêmicos pode ser confundida com abuso infantil.2
O rash típico associado à presença de trombocitopenia sugere púrpura trombocitopênica idiopática ou trombótica, sepse, leucemia e síndrome hemolítico-urêmica. As vasculites que apresentam lesões purpúricas também devem ser consideradas, como poliarterite nodosa, granulomatose de Wegener, crioglobulinemia. A biópsia cutânea é, muitas vezes, fundamental nessa diferenciação.12
As causas mais importantes de abdome agudo, como apendicite e pancreatite aguda, devem ser aventadas devido à presença significativa de dor abdominal no quadro. A PHS e a doença de Berger possuem uma grande semelhança histopatológica. Esta última, no entanto, apresenta curso crônico, evolução lenta e não possui alterações cutâneas.10
No tratamento dos pacientes, é primordial a identificação dos fatores desencadeantes: infecções, alimentos, drogas, vacinas etc. O quadro não responde aos anti-inflamatórios não hormonais e antialérgicos. A base da terapêutica consiste no suporte clínico para alívio dos sintomas. Corticosteroides, imunossupressores, imunoglobulina endovenosa e plasmaférese são reservados para os raros casos de nefrites ou outras manifestações graves.6
A PHS, na maioria dos casos, é autolimitada e benigna, durando em média um mês, mas pode se estender por até dois anos. Nas crianças mais jovens, a doença costuma ser mais breve e com menos recidivas. Os rins devem ser monitorizados com exames periódicos de sangue e urina devido à possibilidade de cronificação da doença. A urina deve ser avaliada diariamente na primeira semana da fase aguda, uma vez por mês por três meses e após a resolução das demais manifestações.4,13
O caso relatado apresentou um curso benigno, necessitando apenas de tratamento sintomático. Até o momento da alta, não foi evidenciado acometimento renal. O paciente foi encaminhado para acompanhamento ambulatorial visando monitoramento de possíveis recidivas ou complicações, que não foram diagnosticadas em consulta posterior, na qual constatou-se ausência de lesões cutâneas e exames laboratoriais de EAS e função renal sem alterações.
No Hospital Universitário Antônio Pedro, de 2012 até 2016, foram realizados 6.704 exames histopatológicos de pele. Desse total, foram achadas 45 vasculites leucocitoclásticas, das quais apenas uma foi concluída, complementarmente à clínica, como púrpura de Henoch-Schönlein.
AGRADECIMENTOS
Agradecemos à Professora Enoí Vilar, do Serviço Patologia, o auxílio na pesquisa dos laudos histopatológicos de vasculite realizados no Hospital Antônio Pedro e as fotografias dos exames do pacient e.
Agradecemos também à mãe do paciente por concordar com o termo de consentimento necessário para a publicação das fotos presentes no relato.
REFERÊNCIAS
1 Alfredo CS, Nunes NA, Len CA, Barbosa CMP, Terreri MTRA, Hilário MOE. Púrpura de Henoch-Schönlein: recorrência e cronicidade. J Pediatr (Rio J). 2007;83(2):177-80.
2 Reamy BV, Williams PM, Lindsay TJ. Henoch-Schönlein purpura. Am Fam Physician. 2009;80(7):697-704.
3 Shetty AK, Desselle BC, Ey JL, Correa H, Galen WK, Gedalia A. Infantile Henoch-Schönlein purpura. Arch Fam Med. 2000;9(6):553-6.
4 Saulsbury FT. Henoch-Schonlein purpura in children. Report of 100 patients and review of the literature. Medicine (Baltimore) 1999;78(6):395-409.
5 Henriques LS, Forte WCN. Púrpura de Henoch-Schönlein [accessed in: Jan 16, 2018]. Available from: http://www.moreirajr.com.br/revistas.asp?fase=r003&id_materia=1613.
6 Silva CAA. Púrpura de Henoch Schonlein [accessed in: Mar 27, 2017]. Available from: https://www.sbp.com.br/arquivo/purpura-de-henoch-schonlein/
7 Mills JA, Michel BA, Bloch DA Calabrese LH, Hunder GG, Arend WP et al. The American College of Rheumatology 1990 criteria for the classification of Henoch-Schönlein purpura. Arthritis Rheumatol. 1990;33(8):1114-21.
8 Weiss PF, Klink AJ, Hexem K, Burnham JM, Leonard MB, Keren R et al. Variation in inpatient therapy and diagnostic evaluation of children with Henoch Schönlein purpura. J Pediatr. 2009;155(6):812-8.
9 Souza MS, Costa JG, Frias ILA. Púrpura de Henoch-Schönlein: relato de caso e revisão da literatura. Resid Pediatr. 2012;2(2):20-7.
10 Kasper DL, Hauser SL, Jameson JL, Fauci AS, Longo DL, Loscalzo J. Medicina interna de Harrison. 19th ed. Porto Alegre: AMGH; 2017. 2 v.
11 Sociedade Europeia de Reumatologia Pediátrica. Púrpura de Henoch-Schönlein [acesso em: 10 jan. 2017]. Disponível em: https://www.printo.it/pediatric-rheumatology/PT/info/8
12 Yang YH, Yu HH, Chiang BL. The diagnosis and classification of Henoch–Schönlein purpura: an updated review. Autoimmun Rev. 2014;13(4-5):355-8.
13 Robson WL, Leung AK. Henoch-Schonlein purpura. Adv Pediatric. 1994;41:163-94.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()