Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 17(1) - Fevereiro 2017
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Pseudo-hipoaldosteronismo tipo 1 como diagnóstico diferencial da hiperplasia adrenal congênita: relato de caso
Pseudohypoaldosteronism type 1 as differential diagnosis of congenital adrenal hyperplasia: case report
Barbara Neffá Lapa e Silva1; Cleo Bragança Cardoso Tammela1; Maria Emmerick Gouveia1; Camila Medeiros de Almeida2; Fernanda Cristina de Carvalho Garcia2; Miguel Luis Graciano3; Valeria Schincariol4; Luciano Abreu de Miranda Pinto5
1. Médica-Residente em Pediatria pela Universidade Federal Fluminense
2. Graduanda em Medicina pela Universidade Federal Fluminense
3. Nefrologista no Hospital Universitário Antônio Pedro/Universidade Federal Fluminense
4. Endocrinologia Pediátrica no Hospital Antônio Pedro/Universidade Federal Fluminense
5. Pediatra no Hospital Universitário Antônio Pedro/Universidade Federal Fluminense e no Hospital Universitário Pedro Ernesto/Universidade do Estado do Rio de Janeiro
Endereço para correspondência:
Instituição: Hospital Universitário Antônio Pedro/Universidade Federal Fluminense
Recebido em: 18.10.2016
Aprovado em: 05.12.2016
Resumo
OBJETIVO: expor os aspectos clínicos e a abordagem do pseudo-hipoaldosteronismo tipo 1, por meio do caso de uma lactente jovem com distúrbio hidroeletrolítico, inicialmente diagnosticada com hiperplasia adrenal congênita.
DESCRIÇÃO DO CASO: este relato descreve o caso de uma lactente jovem, do sexo feminino, que apresentou episódio de desidratação refratária, hiponatremia e hipercalemia. Após extensa investigação, comprovou-se ser um quadro compatível com pseudo-hipoaldosteronismo tipo 1.
DISCUSSÃO: o pseudo-hipoaldosteronismo tipo 1 é uma síndrome rara de resistência aos mineralocorticoides caracterizada clinicamente, no período neonatal, por vômitos, desidratação e ganho pôndero-estatural insatisfatório. Os pacientes acometidos apresentam hiponatremia, hipercalemia e acidose metabólica associada a níveis elevados de aldosterona e renina no plasma. A forma sistêmica do pseudo-hipoaldosteronismo é a mais grave e os sintomas persistem por toda a vida. A forma renal tem apresentação clínica mais leve, com necessidade de suplementação de doses baixas de cloreto de sódio, com regressão dos sintomas no final do primeiro ano de vida.
Palavras-chave: Pseudo-hipoaldosteronismo. Hiperplasia suprarrenal congênita. Hiperpotassemia. Hiponatremia.
Abstract
OBJECTIVE: to expose the clinical aspects and the approach of pseudohypoaldosteronism type 1, through a case of a young infant with electrolyte disturbance initially diagnosed with congenital adrenal hyperplasia.
CASE DESCRIPTION: this report describes a case of a young infant, female, who had an episode of refractory dehydration, hyponatremia and hyperkalemia. After extensive research, it proved to be a pseudohypoaldosteronism with Type 1.
DISCUSSION: pseudohypoaldosteronism type 1 is a rare mineralocorticoid resistance syndrome characterized clinically, in the neonatal period, by vomiting, dehydration and unsatisfactory ponderos-structural gain. The patients affected present with hyponatremia, hyperkalemia and metabolic acidosis associated with elevated plasma aldosterone and renin levels. The systemic form of pseudohypoaldosteronism is the most serious and the symptoms persist for a lifetime. The renal form has a milder clinical presentation, requiring supplementation of low doses of sodium chloride, with regression of symptoms at the end of the first year of life.
Keywords: Pseudohypoaldosteronism. Adrenal hyperplasia, congenital. Hyperkalemia. Hyponatremia.
INTRODUÇÃO
O pseudo-hipoaldosteronismo tipo 1 (PHA1) é uma doença genética rara, determinada por resistência à aldosterona.1 Caracteriza-se por vômitos, desidratação, baixo ganho pôndero-estatural e perda urinária de sal no período neonatal. Crianças afetadas apresentam hiponatremia, hipercalemia, aumento da atividade de renina plasmática e concentrações muito elevadas de aldosterona plasmática.2
Existem duas formas de apresentação: a sistêmica e a renal. A renal, ou tipo 1, é causada por mutações nos receptores de mineralocorticoides (MR) presentes apenas nas células tubulares renais, com apresentação clínica mais leve e necessidade de suplementação de doses baixas de cloreto de sódio.1,2,3 A sistêmica, tipo 2, também conhecida como síndrome de Gordon, está relacionada a alterações no canal de sódio epitelial (ENaC), presente em diversos órgãos e tecidos, como glândulas sudoríparas, salivares, epitélio colônico, rins e pulmões, sendo, portanto, a mais grave, com perda de sal na saliva e no suor, hipercalemia grave e infecções respiratórias de repetição.1 É de herança autossômica recessiva e não apresenta melhora com a idade.1,4,5
Um estudo britânico calculou uma incidência estimada em 1:47.000 para indivíduos acometidos por PHA1. É possível que algumas crianças possam não sobreviver ao episódio inicial da síndrome perdedora de sal, sejam erroneamente classificadas como hiperplasia adrenal congênita (HAC), ou mesmo tenham morte neonatal súbita. Da mesma forma, o PHA do tipo renal pode ser subdiagnosticada, pois normalmente a clínica é mais branda e facilmente tratada com suplementação de sódio.3,6
Além dessas duas formas, uma resistência transitória ao mineralocorticoide também pode ocorrer, por provável maturação anormal do receptor. Essa variante é também chamada de síndrome da hipercalemia infantil.4
Este relato descreve o caso de uma lactente jovem do sexo feminino que apresentou episódio de desidratação refratária, hiponatremia e hipercalemia. Após extensa investigação, comprovou-se ser um quadro compatível com PHA1.
O presente estudo foi aprovado pelo Comitê de Ética em Pesquisa da Universidade Federal Fluminense (CAAE: 0180.0.258.000-09).
DESCRIÇÃO DO CASO
Gemelar feminina, nascida de parto cesáreo, sem intercorrências (mãe não soube informar outros dados), com um mês e 21 dias foi internada com desnutrição proteico-calórica grave, pesando 2,270 kg e desidratação inicialmente atribuída à dieta inadequada, pois não recebeu aleitamento materno e foi alimentada apenas com fórmula infantil em quantidade insatisfatória. No exame físico inicial, suspeitou-se de hipertrofia clitoriana, caracterizada como Pradder I7 e foi aventada hipótese de HAC. Os exames laboratoriais revelaram hiponatremia, hipercalemia e acidose metabólica importantes, mesmo após aporte nutricional adequado. Após constatação da 17-hidroxiprogesterona elevada (521 ng/dl), cortisol e testosterona com níveis plasmáticos normais - r spectivamente 6,08 (VR: 5,0-25 µg/dl) e < 20 (VR: até 73 ng/dl) -, foi iniciada terapia com acetato de fludrocortisona por via oral, em doses que foram aumentadas progressivamente, chegando a 3 mg ao dia. A ultrassonografia abdominal não evidenciava aumento de adrenais. Apesar da otimização terapêutica manteve hiponatremia grave assintomática, elevação do sódio urinário e hipercalemia com alteração eletrocardiográfica, necessitando de terapias específicas para controle eletrolítico, como glicoinsulinoterapia, gluconato de cálcio e resina de troca. Após ganho de peso com aumento da gordura pubiana, foi classificada como estágio zero da escala de Pradder,7 sendo descartada virilização. Diante da não responsividade ao tratamento e ausência de genitália ambígua, o caso foi reavaliado, sendo afastada HAC e suspensa a corticoterapia. A investigação de PHA1 revelou dosagem de aldosterona e renina plasmáticas elevada. Foi iniciada reposição contínua de sódio oral e a paciente evoluiu com bom controle hidroeletrolítico, incluindo normalização dos níveis de potássio.
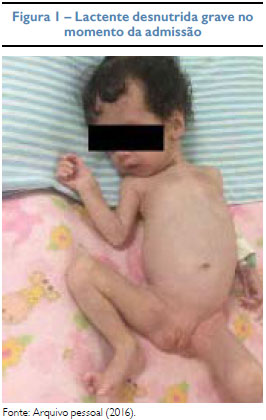
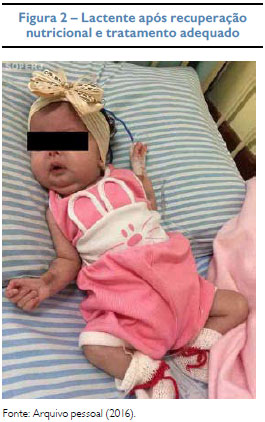
DISCUSSÃO
A desnutrição é uma doença de natureza clínico-social multifatorial. Pode iniciar-se já nos primeiros anos de vida, por interrupção precoce do aleitamento materno, sem adequada suplementação e alimentação complementar. Fatores socioeconômicos, privação alimentar e falta de informação materna podem estar envolvidos. A desnutrição primária está relacionada principalmente aos erros alimentares; e a secundária é causada por comorbidades, como algumas doenças disabsortivas. A desnutrição apresenta-se em um espectro de manifestações clínicas e laboratoriais. Em relação aos distúrbios eletrolíticos destaca-se a hipocalemia e hipomagnesemia.8 A perpetuação da desnutrição, apesar do tratamento correto, e a hipercalemia, presente desde o início do quadro, mostraram a necessidade de uma investigação mais detalhada.
Diante de um lactente jovem com desidratação, baixo peso, hiponatremia e hipercalemia graves, a primeira hipótese aventada é HAC, por ser a causa mais comum do quadro descrito. Entretanto, após descartar sinais de virilização com ganho ponderal e da ausência de resposta ao acetato de fludrocortisona, investigou-se outras etiologias. O principal diagnóstico diferencial deve ser feito com o PHA1, uma vez que sua apresentação clínica é similar à HAC.1 O diagnóstico é realizado pela dosagem de aldosterona e renina elevada, e o tratamento baseia-se na reposição oral de sódio e correção do potássio com resina de troca.1,5
O PHA caracteriza-se por resistência dos receptores teciduais aos mineralocorticoides. A aldosterona sintetizada pelo córtex da suprarrenal faz sua ação por meio dos receptores MR e ENaC. Dentre seus efeitos, promove reabsorção de sódio e secreção de potássio, com consequências no sistema cardiovascular, além de exercer efeitos diretos importantes sobre o sistema nervoso central e sobre o balanço energético.1
A doença apresenta-se em duas formas: sistêmica, com manifestações clínicas mais graves; e a forma renal, de apresentação clínica mais branda e com possibilidade inclusive de remissão dos sintomas.2,4,9
No caso apresentado, a lactente apresentava distúrbios hidroeletrolíticos na ausência de desidratação grave ou infecções respiratórias de repetição, levando à suspeita clínica da forma renal do PHA. Além disso, houve melhora do quadro com baixa suplementação de cloreto de sódio.
A diferenciação entre as formas de apresentação do PHA se faz necessária para o seguimento e tratamento dos pacientes. Na forma renal, há a possibilidade de remissão dos sintomas e normalização eletrolítica no final do primeiro ano de vida, podendo o tratamento ser interrompido por volta dos 18-24 meses. As crianças mais velhas são assintomáticas, com desenvolvimento psicomotor normal, embora possam evoluir em percentis inferiores da curva de crescimento.1,2,4,9 Uma possível explicação para esta melhora seria o aumento da concentração de sal na dieta de lactentes quando da transição de aleitamento materno exclusivo para dieta mista, sugerindo uma interação genético-ambiental.2 Com uma dieta de baixa ingestão de sódio, o neonato e o lactente seriam dependentes de uma ativação máxima do sistema renina-angiotensina-aldosterona e a haploinsuficiência dos MR resultaria em depleção de volume e hipercalemia.9
Para o PHA1 sistêmico, normalmente são usadas elevadas doses de cloreto de sódio por toda a vida. Em casos extremos, pode ser necessário gastrostomia para fornecer as quantidades ideais de suplementação do sal e diálise peritoneal na hipercalemia grave.1
CONCLUSÃO
Devido à raridade, o PHA1 não costuma ser considerado no diagnóstico diferencial de lactentes com perda de sal e hipercalemia. Portanto, uma melhor compreensão da doença é relevante para o correto tratamento de síndromes perdedoras de sal e para abrir novas perspectivas sobre a abordagem da regulação hidroeletrolítica nesta doença.
REFERÊNCIAS
1 Miranda M, Fontoura M. Pseudohipoaldosteronismo tipo 1 na criança. Arq de Med. 2013;27(6):265-71.
2 Fernandes-Rosa FL, Antonini SRR. Resistência aos mineralocorticoides: pseudo-hipoaldosteronismo tipo 1. Arq Bras Endocrinol Metab. 2007;51(3):373-81.
3 Amin N, Alvi NS, Barth JH, Field HP, Finlay E, Tyerman K, Frazer S, Savill G, Wright NP, Makaya T, Mushtaq T. Pseudohypoaldosteronism type 1: clinical features and management in infancy. Endocrinol Diabetes Metab Case Rep. 2013;2013:130010.
4 Sperling MA. Endocrinologia pediátrica. 4. ed. United States: Elsevier; 2015. p. 165-66.
5 Bangash AS, Ali NF, Sami S, Iqbal M. Pseudohypoaldosteronism type-1: a rare cause of hyperkalemia in neonates. J Pak Med Assoc. 2014;64(4):484-6.
6 Güran T, Degimenci S, Bulut IK, Aysun S, Felix GR, Ömer G. Critical points in the management of pseudohypoaldosteronism type 1. J Clin Res Pediatr Endocrinol. 2011;3(2):98-100.
7 Damiani D, Setian N, Kuperman H, Manna TD, Dichtchekenian V. Genitália ambígua: diagnóstico diferencial e conduta. Arq Bras Endocrinol Metab. 2001;45(1):37-47.
8 Brasil. Ministério da Saúde. Manual de atendimento da criança com desnutrição grave em nível hospitalar. Brasília: MS; 2005 [acesso em 27 dez 2016]. Disponível em: http://bvsms.saude.gov.br/bvs/publicacoes/manual_desnutricao_criancas.pdf.
9 Geeler DS. Mineralocorticoid resistance. Clin Endocrinol. 2005;62(5):513-20.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()