Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 16(3) - Outubro 2016
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
RELATOS DE CASO
Polineuropatia desmielinizante inflamatória crônica na infância
Chronic inflammatory demyelinating polyneuropathy in infancy
Ayla Cristina Bernardes da Silva1; Lívia Batista Corrêa1; Douglas Covre Stocco Renato Abreu Ribeiro1; Isabela Felício2; Marcio Moacyr Vasconcelos3
1. Internos, Serviço de Pediatria, Hospital Universitário Antônio Pedro (HUAP), Universidade Federal Fluminense (UFF), Niterói, RJ
2. Médica residente, Serviço de Neuropediatria, Hospital Universitário Antônio Pedro (HUAP), Universidade Federal Fluminense (UFF), Niterói, RJ
3. Professor adjunto, Hospital Universitário Antônio Pedro (HUAP), Universidade Federal Fluminense (UFF), Niterói, RJ
Endereço para correspondência:
Ayla Cristina Bernardes da Silva
Rua Alvares de Azevedo, 130/1002B
24220-021, Niterói, RJ
Telefone: (21) 41266676
E-mail: ayla.bernardes@yahoo.com.br
Recebimento 18.10.2015
Aprovaçao 23.11.2015
Resumo
OBJETIVO: Relatar um caso de polineuropatia desmielinizante inflamatória crônica na infância (PDIC).
DESCRIÇÃO DO CASO: Paciente do sexo feminino, 5 anos, internada com quadro progressivo de dor e paresia em membros inferiores, com evolução dos primeiros sintomas há 3 meses, com perda de deambulação e de se manter em posição ortostática, apresentando diversas recidivas em meses, após tratada.
DISCUSSÃO: A PDIC é uma doença que envolve os membros proximal e distal, com achados eletroneuromiográficos de desmielinização multifocal de nervos somáticos. Os sintomas iniciais são fraqueza muscular simétrica e progressiva, em episódios, com tato comprometido e reflexos osteotendinosos comprometidos. É uma doença recidivante, crônica e progressiva, com quadro similar à Síndrome de GuillainBarré (SGB). O presente relato permite a discussão da avaliação, manifestações clínicas, diagnóstico, seguimento e estratégia terapêutica para a Polineuropatia Desmielinizante Inflamatória Crônica.
Palavras-chave: GuillainBarré, polineuropatias, criança, relatos de caso.
Abstract
OBJECTIVE: We aim to report a case of chronic inflammatory demyelinating Polyneuropathy (PDIC).
CASE DESCRIPTION: Patient female, 5 years-old, hospitalized, presenting progressive pain and paresis in lower members, ambulation loss and incapacity of staying in orthostatic position, with first symptoms evolution in 3 months. After treatment, the patient presents several relapses in months.
DISCUSSION: PDIC is a disease which evolves proximal and distal members parts, with electroneuromyographycs findings of somatic nerves multifocal demyelination. The first symptoms are symmetric and progressive muscular weakness, in episodes, touch and osteotendinous reflexes damage. PDIC is a relapsing disease, chronic and progressive, similar to GuillainBarré syndrome (SGB). The present case report allows discussion about evaluation, clinical manifestations, diagnosis, follow-up and therapeutic strategy in chronic inflammatory demyelinating polyneuropathy.
Keywords: GuillainBarré, polyneuropathies, child, case reports.
INTRODUÇÃO
A polineuropatia desmielinizante inflamatória crônica (PDIC) é uma polineuropatia com progressão de 2 meses ou mais, que envolve os membros proximal e distal, com achados eletroneuromiográficos indicativos de desmielinização multifocal dos nervos somáticos e, em parte dos casos, pode ser documentada por meio de biópsia de nervo.
Trata-se de doença com prevalência de 1/200.000 nas crianças e de 17/100.000 nos adultos. As principais manifestações incluem fraqueza muscular simétrica e progressiva, com recuperação parcial ou total entre agudizações, associadas a tato comprometido e ausência ou diminuição de reflexos osteotendinosos.
A doença é recidivante em 30% dos casos; crônica e progressiva em 60%; e monofásica, geralmente com recuperação completa permanente, em 10% Apresenta quadro clínico, patológico e laboratorial semelhante à Síndrome de GuillainBarré (SGB), mas, na maioria das vezes, possui evolução crônica e responde satisfatoriamente à terapêutica com glicocorticoides. O tratamento é realizado com imunoglobulina humana endovenosa sendo os corticoides e os imunossupressores reservados para a doença refratária.
DESCRIÇÃO DO CASO
Paciente IHL, sexo feminino, 6 anos de idade, admitida no serviço de Emergência Pediátrica do Hospital Universitário Antônio Pedro, Niterói, Rio de Janeiro. História relatada pela mãe descreve queixa de algia e perda da força muscular em membro inferior esquerdo (MIE) há 3 meses, com evolução após 2 meses para membro inferior direito (MID), dificuldade de deambulação e de manter-se em posição ortostática após várias quedas. Nega envolvimento de esfíncteres, alterações na fala, na deglutição ou alterações visuais, bem como cefaleia, vômitos e outros sintomas. Cavidade oral e otoscopia não apresentaram alterações. Nesse período, realizou fisioterapia, sem melhora do quadro.
Além dessas, coletamos as seguintes informações:
(i) História patológica pregressa - sem história de infecção de vias aéreas ou gastroenterite prévia. Nega doenças graves, cirurgias ou internações prévias.
(ii) História gestacional e do parto - peso de 3.350 g; comprimento de 51 cm; perímetro cefálico de 36 cm; apgar de 8/9;alta após 3 dias.
(iii) Desenvolvimento - deambulou com 13 meses e falou com 1 ano.
(iv) História vacinal sem história de vacinação nos últimos meses (sic).
(v) Exame físico perímetro cefálico de 50 cm; pressão arterial de 110x50 mmHg;frequência cardíaca de 96 bpm; frequência respiratória de 22 irpm. Nuca livre. Ritmo cardíaco regular em 2 tempos, bulhas normofonéticas, sem sopros. Ausculta pulmonar de murmúrios vesiculares universalmente audíveis, sem ruídos adventícios. Abdome normal. Genitálias e membros sem alterações.
Após avaliação do setor, foi pedido parecer da Neurologia, que, ao exame, evidenciou paciente alerta, cooperativa e respostas adequadas; teste de nervos de pares cranianos com pupilas em 3 mm e fotorreativas, visão normal e fundoscopia sem papiledema; palato e língua normais; exame motor com hipotonia muscular difusa, reflexos tendíneos periféricos abolidos nos membros inferiores (MMII) e diminuídos nos membros superiores (MMSS); força muscular de 3/5 nos MMSS e de 2/5 nos MMII; exame sensitivo sem alteração. Ao exame de coordenação dedo-nariz, não foi observada ataxia, enquanto à marcha, a paciente não consegue ficar em posição ortostática.
Além disso, realizou-se o estudo da condução nervosa, que evidenciou paralisia flácida aguda, ascendente, bilateral e simétrica de evolução subaguda, associada a exame sensitivo normal, com quadro sugestivo de SGB. Também foram solicitados tomografia computadorizada de crânio (sem alteração), punção lombar, CK, eletrólitos e fosfatase alcalina, evidenciando apenas de alteração um aumento nos miligramas, com valor de 12,5 mg/dL.
A paciente foi tratada com imunoglobulina humana endovenosa na dose de 2g/kg por 5 dias, com melhora do quadro, e recebeu alta. Após 1 ano da data da primeira internação, apresentou as mesmas queixas em mais 3 episódios, sendo também internada. Realizou-se estudo do líquido cefalorraquidiano, que apresentou aspecto levemente hemorrágico, glicose de 56 mg/dL e ausência de células material, com LDH de 15 U/L.
Outro estudo da neurocondução foi procedido na terceira internação (tabela 1), 4 meses após a primeira, sendo compatível com polineuropatia sensitivo-motora desmielinizante. Assim, chegou-se ao diagnóstico de PDIC.
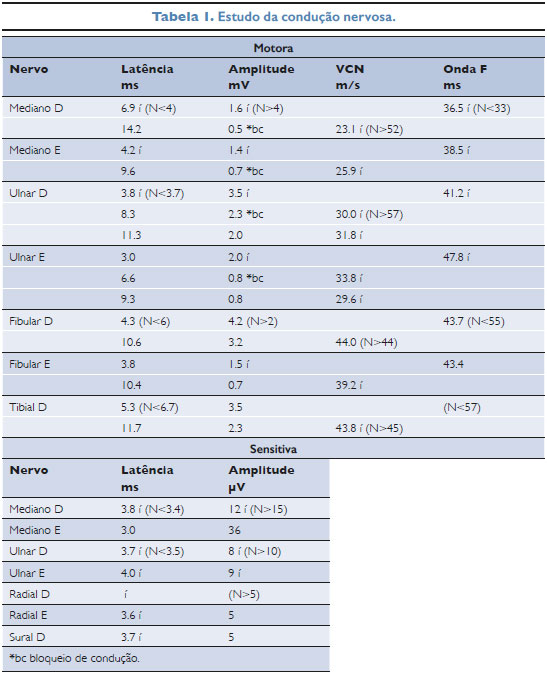
Na última internação, pela Neuropediatria, foi tratada com imunoglobulina humana endovenosa na dose de 1g/kg em infusão lenta por 2 dias, com 3 picos febris (38,5°C, 38,1°C e 38,1°C) durante a administração. Apresentou melhora e recebeu alta 3 dias após. O controle ambulatorial mensal demonstrou exames neurológicos normais, regressão do quadro, sem limitar atividades habituais e com indicação de permanência da fisioterapia e atendimento no serviço.
DISCUSSÃO
A PDIC é uma doença autoimune caracterizada por fraqueza em músculos proximais e distais dos membros, a qual piora progressivamente por mais de 2 meses. A prevalência em crianças ainda é menor em relação aos adultos; talvez por isso, ainda existam poucos estudos sobre a real prevalência e o comportamento na população infantil em comparação à adulta. 1
Caracteriza-se por comprometimento periférico ascendente, progressivo e geralmente simétrico, em que as manifestações motoras predominam sobre as sensoriais. Com frequência, os sintomas iniciais constituem formigamento e "sensações de alfinetadas e agulhadas" nos pés, podendo ser associados à lombalgia aguda. Além disso, há perda de força dos membros inferiores, perda do controle esfincteriano, comprometimento de pares cranianos e diminuição dos reflexos tendinosos profundos. Os sinais que devem chamar a atenção do pediatra geral são aqueles que comprometem a ventilação, a deglutição e os movimentos oculares.2,7 Propriedades distintivas na criança incluem progressão subaguda em menos de 2 meses, envolvimento do sistema do motor predominante nas extremidades inferiores e melhoria acentuada na resposta a terapia de modulação imunitária.1,4
O processo fisiopatológico básico da PDIC, no que se refere à desmielinização inflamatória, parece envolver fatores imunológicos. Os mecanismos imunes celular e humoral têm, provavelmente, um papel no desenvolvimento da doença. Alguns pontos críticos da doença permanecem enigmáticos, incluindo a natureza e o local da resposta imune, assim como os fatores do hospedeiro que permitem o desenvolvimento da PDIC. Em 2/3 dos casos, uma infecção respiratória aguda ou gastrointestinal precede o início em 2 a 3 semanas.2,4
São diagnósticos diferenciais a serem considerados: neuroencefalopatias mitocondriais hereditárias, Doença de CharcotMarieTooth, adrenoleucodistrofia, adrenomieloneuropatia, leucodistrofia metacromática, Doença de Krabbe, distúrbios adquiridos, como, por exemplo, hipotireoidismo ou hipertireoidismo, e distúrbios causados por exposição a agentes tóxicos, como, por exemplo, a colchicina e cloroquina.3,5,8
Não há qualquer teste específico para o diagnóstico, sendo realizado pelas características clínicas e pela alteração do líquido cefalorraquidiano (proteína elevada mais que o dobro do limite superior do normal, nível de glicose é normal e sem pleocitose). Os exames eletrodiagnósticos cuidadosos geralmente podem identificar, pelo menos, anormalidades leves nos estágios iniciais.7 Todas as circunstâncias restantes que se assemelham à SGB devem também ser excluídas.1,2,5,6
As alternativas terapêuticas incluem corticosteroides, imunoglobulina humana endovenosa em altas doses e, eventualmente, plasmaférese.8-10 Embora a terapia de manutenção seja praticada rotineiramente, a necessidade de continuar terapias de longo prazo deve ser revista periodicamente para evitar o excesso de tratamento.5,8 O tratamento precoce e eficaz antes que ocorra a degeneração axonal significativa é o mais importante fator que determina o resultado do tratamento.11,1 Crianças apresentam melhores respostas à terapia imunológica, mas têm maior tendência à recaída.1
REFERÊNCIAS BIBLIOGRÁFICAS
1. Jo HY, Park MG, Kim DS, Nam SO, Park KH. Chronic inflammatory demyelinating polyradiculoneuropathy in children: characterized by subacute, predominantly motor dominant polyneuropathy with a favorable response to the treatment. Acta Neurol Scand 2010; 121: 342347.
2. Bolan RS, Bó KD, Vargas FR, Moretti GRF, Almeida LP, Almeida GKP, et al. Síndrome de Guillain-Barré. Rev AMRIGS 2007; 51 (1): 5861.
3. Carbot RC, Scully RE, Mark EJ, McNeely WF, Ebeling SH, Phillips LD. Weekly clinicopathological exercises: case 131998. N Engl J Med 1998; 338(17): 12121219.
4. Torricelli RE. Síndrome de Guillain Barre en Pediatría. Medicina 2009; 69 (1): 8491.
5. Dalakas MC. Advances in the diagnosis, pathogenesis and treatment of CIDP. Nat Rev Neurol 2011; 7: 507517.
6. SchneiderHohendorf T, Schwab N, Üçeyler N, Göbel K, Sommer C, Wiendl H. CD8+ Tcell immunity in chronic inflammatory demyelinating polyradiculoneuropathy. Neurol 2012 ; 78: 402408.
7. Dyck PJ. Atypical varieties of chronic inflammatory demyelinating neuropathies. Lancet 2000; 355: 12931294.
8. Sousa EA. Chronic inflammatory demyelinating polyneuropathy: diagnosis and management. Exp Rev Clin Immunol 2010; 6(3): 373.
9. Wrobel CJ, Watson D. Plasmapheresis in chronic demyelinating polyneuropathy. N Engl J Med 1992; 326(16): 10891090.
10. Börü ÜT, Erdogan H, Alp R, Tasdemir M, Yıldırım S, Bilgiç A, et al. Treatment of chronic inflammatory demyelinating poly-neuropathy with high dose intravenous methylprednisolone monthly for five years: 10Year follow up. Clin Neurol Neurosurg 2014; 118: 8993.
11. Chan YC, WilderSmith E. Predicting treatment response in chronic, acquired demyelinating neuropathies. Exp Rev Neurother 2006; 6(10): 1545.
12. Sinno DD, Darras BT, Yamout BI, Rebeiz JG, Mikati MA. Motor variant of chronic inflammatory demyelinating polyneuropathy in a child. Pediatr Neurol 2008; 38(6): 426429.
13. Kam C, Balaratnam MS, Purves A, Mills IKR, RiordanEva P, Pollock S, et al. Canomad presenting without ophthalmoplegia and responding to intravenous immunoglobulin. Muscle Nerve 2011; 44: 829833.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()