Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 16(3) - Outubro 2016
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
RELATOS DE CASO
Doença de Gaucher como diagnóstico diferencial de esplenomegalia
Gaucher disease in differential diagnosis of splenomegaly
Tânia de Cássia Moreira Soares1; Jamille Fernandes Lula2; Leandro de Freitas Teles3; Daniele Mesquita de Brito4; Silvio Fernando Guimaraes de Carvalho5
1. Mestrado Analista Universitário da Saúde do Hospital Universitário Clemente de Faria da Universidade Estadual de Montes Claros (HUCF-UNIMONTES)
2. Doutorado Analista Universitário da Saúde do Hospital Universitário Clemente de Faria da Universidade Estadual de Montes Claros (HUCF-UNIMONTES)
3. Doutorando em Ciências da Saúde da Universidade Estadual de Montes Claros Analista Universitário da Saúde do HUCF-UNIMONTES, Analista de Hematologia e Hemoterapia da Fundaçao Hemominas
4. Graduanda de Medicina da UNIMONTES
5. Doutorado Médico Pediatra e responsável pelo serviço do Centro de Pesquisa em Doenças Infectoparasitárias do HUCF-UNIMONTES
Endereço para correspondência:
Tânia de Cássia Moreira Soares
Hosp. Universitário Clemente de Faria
Avenida Cula Mangabeira, 432 Santo Expedito
39401-001 - Montes Claros-MG
Tel.: (38) 3224-8378
E-mail: taniamoreiracs@hotmail.com
Recebimento 25.03.2016
Aprovaçao 15.05.2016
Resumo
OBJETIVO: Apresentar a doença de Gaucher como diagnóstico diferencial das esplenomegalias na infância.
DESCRIÇÃO DO CASO: Este artigo relata um caso de criança do sexo feminino, seis anos, com anemia e esplenomegalia, equimoses esporádicas, episódios de dores ósseas e abdominais de repetição. Realizou-se o diagnóstico por meio da atividade diminuída da Beta-glicosidase. Após nove meses de tratamento com imiglucerase, a criança apresentou desenvolvimento pôndero-estatural satisfatório, e os exames hematológicos e as vísceras estavam dentro dos limites da normalidade.
DISCUSSÃO: A doença de Gaucher é uma doença multissistêmica e faz parte do diagnóstico diferencial das esplenomegalias na infância, sendo considerada a mais prevalente entre mais de 50 tipos de doenças de estoque lisossômico até então descritas. Ocorre o acúmulo de glicocerebrosídeos nos macrófagos teciduais, alterando o funcionamento normal desses órgãos e tecidos, o que pode causar danos irreversíveis. Os pacientes com a doença de Gaucher têm apresentações clínicas variáveis, o que nem sempre se correlaciona com genótipos específicos. Considerando o fato de a esplenomegalia ser comum a várias patologias, faz-se necessária a inclusão da doença de Gaucher em seu vasto diagnóstico diferencial. Apesar de a doença de Gaucher ser uma condição infrequente, apresenta um quadro clínico muito semelhante ao de doenças de elevada prevalência, necessitando de um diagnóstico precoce e específico, com acompanhamento multidisciplinar, para obter o controle clínico adequado e consequente menor morbidade.
Palavras-chave: esfingolipidoses; doença de Gaucher; esplenomegalia.
Abstract
OBJECTIVE: We aim to present Gaucher disease in the differential diagnosis of esplenomegalias in childhood.
CASE DESCRIPTION: This article reports a case of female child of six years-old, with anemia and splenomegaly, bruises sporadic, episodes of bone and abdominal pain of repetition. She carried out the diagnosis by decreased activity of Betaglicosidase. After nine months of treatment with imiglucerase, the child presented weight and height satisfactory development, and hematology and viscera were within normal limits.
DISCUSSION: Gaucher disease is a multisystem disease and is part of the differential diagnosis of splenomegaly in childhood. It is the most prevalent among more than 50 types of stock lysosomal diseases described. Glucocerebrosides accumulate in tissue macrophages, altering normal functions of these organs and tissues, and causing irreversible damages. Patients with Gaucher disease have variable clinical presentation and do not always correlate with specific genotypes. Considering the fact splenomegaly is common to many diseases, it is necessary the inclusion of Gaucher disease in its broad differential diagnosis. Although Gaucher disease is an uncommon condition, it presents a clinical picture very similar to high prevalence diseases, requiring an early and specific diagnosis, with multidisciplinary follow-up, in order to obtain proper clinical management and consequently lower morbidity.
Keywords: sphingolipidoses; Gaucher disease; splenomegaly.
INTRODUÇÃO
Este artigo tem como objetivo apresentar a doença de Gaucher como diagnóstico diferencial das esplenomegalias na infância. Apesar das manifestações clínico-laboratoriais iniciarem na infância, o diagnóstico, na maioria dos casos, é tardio, levando ao prejuízo no tratamento precoce ou desenvolvendo complicações irreversíveis.1,2
RELATO DE CASO
Criança do sexo feminino, seis anos, admitida no ambulatório de Hematologia da Universidade Estadual de Montes Claros (Unimontes), com anemia e esplenomegalia a esclarecer. Relato de anemia persistente há três anos, equimoses esporádicas, episódios de dores ósseas e abdominais de repetição. A criança esteve internada, aos quatro anos de idade, com quadro de febre e esplenomegalia, recebendo alta com o diagnóstico provável de mononucleose infecciosa.
Um mês antes da consulta hematológica, internou-se com quadro de febre, dor em membro inferior direito e radiografia sugestiva de osteomielite, sendo submetida à antibioticoterapia com remissão do quadro febril. História familiar negativa para patologias hematológicas.
Ao exame físico, apresentava-se com hipodesenvolvimento pôndero-estatural, hipocorada, hepatoesplenomegalia moderadas, afebril, anictérica, ausência de linfadenomegalias, exame neurológico sem alterações. Os exames laboratoriais revelaram anemia (hemoglobina 10,4 g/dL), trombocitopenia (93x103/mL), leucócitos sem alterações, transaminases levemente alteradas, ferritina, ácido fólico, vitamina B12, desidrogenase lática, haptoglobina, eletroforese de hemoglobina, pesquisa molecular de α-talassemia, teste de fragilidade osmótica, coombs, bilirrubinas e γ-gt apresentavam dentro dos limites da normalidade. Sorologias para toxoplasmose, rubéola, Epstein Baar, citomegalovírus, hepatites B e C, sífilis foram negativas.
A radiografia de fêmur direito apresentava lesão osteolítica permeativa na metáfise distal; a radiografia de mão para avaliar a idade óssea era compatível com a idade de 3 anos e 6 meses; a ressonância magnética de coluna lombar e fêmures com alterações de sinal difusas e dos espaços medulares das diáfises e metáfises (Figura 1); o mielograma apresentava-se com células reticulares sugestivas de células de Gaucher.
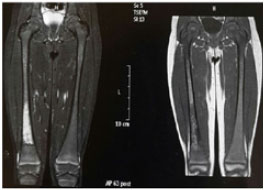
Figura 1
A atividade da Beta-glicosidase em leucócitos 1,4 nmol/h/mg de proteínas (valor normal: 8,0-35,6 nmol/h/mg) e a atividade da Quitotriosidase 8219,0 nmol/h/mL (valor normal: 1,7-175,7 nmol/mL). Após nove meses de tratamento com imiglucerase (60UI/kg intravenosa de 15 em 15 dias), a criança cresceu 7 cm e ganhou 2 kg, os exames hematológicos e as vísceras encontravam-se dentro dos limites da normalidade.
DISCUSSÃO
A doença de Gaucher, descoberta pelo médico Philippe Charles Ernest Gaucher em 1882, é multissistêmica, sendo a mais prevalente entre mais de 50 tipos de doenças de estoque lisossômico até então descritas. Tornou-se um padrão para a descrição da variabilidade fenotípica dessas doenças; apresenta uma herança autossômica recessiva; e o risco de desenvolver a doença aumenta com a consanguinidade da família. A sua frequência é diferente em variadas populações, com maior prevalência em judeus Ashkenazi 1:450 nascidos vivos. No entanto, a prevalência global estimada de doença sintomática é menor, ocorrendo em, aproximadamente, 1 em 40.000-100.000 nascidos vivos. Devido à baixa frequência, é incluída nas chamadas enfermidades órfãs e, possivelmente, afeta menos de 10.000 pessoas no mundo.3
É uma patologia causada por mutações no gene GBA (1q21) e mais de 200 alelos mutantes foram identificados. Esse gene que codifica a enzima beta-glicosidase ácida, também conhecida como glicocerebrosidase, uma enzima lisossômica que catalisa a hidrólise de glicocerebrosídeo em ceramida e glicose. A deficiência da glicocerebrosidase induz a um acúmulo de glicolipídeos não digeridos (substratos) e leva a alterações histológicas, especialmente evidentes nos órgãos que são ricos em elementos do sistema imune monocítico-fagocitário (baço, fígado e medula óssea).3,4
Os pacientes com a doença de Gaucher têm apresentações clínicas variáveis e nem sempre se correlacionam com genótipos específicos, o que indica a existência de outros fatores, genéticos ou ambientais, influenciando o fenótipo. Em casos mais graves, essa doença pode afetar os pulmões, os rins e o sistema nervoso central. Esse acúmulo altera o funcionamento normal desses órgãos e tecidos, o que pode causar danos irreversíveis.4,5
Os glicocerebrosídeos, que se acumulam em órgãos viscerais, resultam de membranas de células sanguíneas em processo de envelhecimento, as quais contêm glicoesfingolípideos, incluindo leucócitos, eritrócitos e plaquetas, levando ao desenvolvimento de hepatomegalia, esplenomegalia, expansão da medula óssea e ativação e proliferação de macrófagos com subsequente produção de numerosas citocinas e quimiocinas. Os efeitos pró e anti-inflamatórios decorrentes dessas citocinas provocarão danos teciduais que podem se tornar irreversíveis. O mecanismo exato pelo qual ocorre essa ativação de macrófagos não está bem definido.6
Essa deficiência enzimática não é completa, independentemente da gravidade, os pacientes irão apresentar certa quantidade residual de atividade enzimática em todos os tipos de células nucleadas; a ausência enzimática completa em seres humanos ou em cobaias é letal em prematuros ou no período neonatal. As manifestações clínicas ou fenotípicas da doença de Gaucher dependerão do grau de deficiência da enzima. A presença e a gravidade dos sintomas neurológicos definirão os três tipos clínicos.
Aproximadamente 95% dos pacientes apresentam a doença de Gaucher do tipo 1 ou doença não neuropática sem envolvimento neurológico e com início durante a infância ou na idade adulta, bem como apresentam esplenomegalia progressiva como uma de suas primeiras manifestações, achado comum a todos os casos. Além disso, o baço pode atingir até 20% do peso corporal.
Crianças com esplenomegalia maciça são de baixa estatura por causa do gasto de energia requerida pelo órgão afetado. Ocorre, ainda, hepatomegalia com ou sem testes de função hepática alterados, expansão da medula óssea, fadiga crônica, anemia e trombocitopenia. Raramente, outros órgãos podem ser acometidos. O acometimento ósseo pode apresentar em crises ósseas agudas secundárias necrose avascular aguda, dor óssea crônica, infarto medular, osteopenia, osteoporose, edema local, fraturas patológicas ou falha de crescimento. A maioria dos pacientes tem evidência radiológica de envolvimento do esqueleto, incluindo uma deformidade distal do fêmur semelhante ao frasco de Erlenmeyer, e a ressonância magnética é mais sensível do que a tomografia computadorizada para demonstrar a extensão das anormalidades em pacientes com doença de Gaucher e pode ter um valor prognóstico.7,8,10-12
A doença tipo 2 tem uma progressão neurológica rápida com envolvimento visceral acentuado e morte nos primeiros dois anos de vida. As crianças apresentam aumento do tônus muscular, estrabismo, falha de crescimento e típico estridor laríngeo devido a laringoespasmos. A rápida degeneração psicomotora leva à morte, geralmente devido ao comprometimento respiratório progressivo. Os pacientes com a doença de Gaucher tipo 3 são diagnosticados na infância ou na adolescência. Apresentam envolvimento ósseo, visceral e neurológico; porém, menos acentuado que o tipo 2 e pode atingir a vida adulta.7,8,10-12.
O diagnóstico clínico da doença de Gaucher pode ser confirmado pela análise da atividade da beta-glicosidase ácida. Além disso, ao contrário do diagnóstico enzimático, a análise da mutação tem um valor no que se refere ao prognóstico da doença.9 A terapia de reposição enzimática (TRE) eficiente foi desenvolvida e tornou-se um exemplo para a intervenção terapêutica em genética humana. A TRE é o tratamento padrão para pacientes portadores da doença de Gaucher tipo 1 e 3. A dose da enzima é estabelecida para cada paciente de acordo com o peso corporal e as manifestações clínicas.6
Considerando o fato de a esple- nomegalia ser comum a várias patologias, faz-se necessária a inclusão da doença de Gaucher em seu vasto diagnóstico diferencial. Chama-se a atenção para o cuidado na avaliação clínica precisa e cautelosa, e as decisões invasivas como a esplenectomia diagnóstica devem ser evitadas sempre que possível. Apesar de a doença de Gaucher ser uma condição infrequente, considerada doença órfã, apresenta um quadro clínico muito semelhante ao de doenças de elevada prevalência, necessitando de um diagnóstico precoce e específico, com acompanhamento multidisciplinar para obter o controle clínico adequado e consequente menor morbidade.
REFERÊNCIAS BIBLIOGRÁFICAS
1. Gaucher registry: treating & researching Gaucher disease. Disponível em: <https://www.registrynxt.com/Gaucher/Pages/Home>.
2. Pozo AL, Godfrey EM, Bowles KM. Splenomegaly: investigation, diagnosis and management. Blood rev 2009; 23(3): 105-11.
3. Grabowski GA. Gaucher disease and other storage disorders. Hematology, The Education Program of the American Society of Hematology. 2012; 2012: 13-8.
4. Rozenberg R, Araujo FT, Fox DC, Aranda P, Nonino A, Micheletti C, et al. High frequency of mutation G377S in Brazilian type 3 Gaucher disease patients. Braz J Med Biol Res 2006; 39(9): 1171-9.
5. Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008; 29(5): 567-83.
6. Sobreira EAP, Bruniera P. Avaliação de dois anos de tratamento da doença de Gaucher tipo 1 com terapia de reposição enzimática em pacientes do estado de São Paulo, Brasil. Rev Bras de Hemat e Hemoter 2008; 30: 193-201.
7. Nagral A. Gaucher disease. J Clin Exp Hepatol 2014; 4(1): 37-50.
8. Di Rocco M, Andria G, Deodato F, Giona F, Micalizzi C, Pession A. Early diagnosis of Gaucher disease in pediatric patients: proposal for a diagnostic algorithm. Pediatr Blood Cancer 2014 ; 61(11): 1905-9.
9. Rosenthal DI, Scott JA, Barranger J, Mankin HJ, Saini S, Brady TJ, et al. Evaluation of Gaucher disease using magnetic resonance imaging. J Bone Joint Surg Am 1986; 68(6): 802-8.
10. Mistry PK, Weinreb NJ, Kaplan P, Cole JA, Gwosdow AR, Hangartner T. Osteopenia in Gaucher disease develops early in life: response to imiglucerase enzyme therapy in children, adolescents and adults. Blood Cells Mol Dis 2011; 46(1): 66-72.
11. Poll LW, Cox ML, Godehardt E, Steinhof V, vom Dahl S. Whole body MRI in type I Gaucher patients: evaluation of skeletal involvement. Blood Cells Mol Dis 2011; 46(1): 53-9.
12. Thomas AS, Mehta AB, Hughes DA. Diagnosing Gaucher disease: an on-going need for increased awareness amongst hematologists. Blood Cells Mol Dis 2013; 50(3): 212-7.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()