Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 25(3) - Setembro 2025
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Síndrome de Wolf-Hirschhorn: um relato de caso
Wolf-Hirschhorn syndrome: a case report
Ana Luíza Nardelli-Kühl1; Samantha Lopes1; Simone Cristina Padilha Stadnick1,2
DOI:10.31365/issn.2595-1769.v25i3p124-128
1. Centro Universitário para o Desenvolvimento do Alto Vale do Itajaí - UNIDAVI, Núcleo de Pesquisa em Ciências Médicas - NPC Med, Faculdade de Medicina - Rio do Sul-SC, Brasil
2. Hospital Regional do Alto Vale, Emergência pediátrica - Rio do Sul-SC, Brasil
Endereço para correspondência:
Instituição: Faculdade de Medicina, Rio do Sul, SC, Brasil
Recebido em: 06/10/2024
Aprovado em: 17/02/2025
Resumo
A síndrome de Wolf-Hirschhorn (SWH) é causada por uma deleção genética do braço curto do cromossomo 4 (4p16.3). Possui incidência de 1:50.000 nascidos vivos e acometimento na proporção de 2:1 entre mulheres e homens. A extensão da deleção cromossômica determina as variações fenotípicas, que podem envolver alterações craniofaciais, crises epilépticas, atraso do desenvolvimento neuropsicomotor, microcefalia, hipotonia e malformações de múltiplos sistemas, principalmente o cardíaco. Este estudo irá descrever um caso de SWH em uma criança, feminina, diagnosticada por meio do estudo genético de microarranjo de DNA. Dentre as manifestações da anomalia genética, a criança apresentou o sinal craniofacial típico, crises convulsivas parciais e generalizadas, atraso grave do desenvolvimento neuropsicomotor, microcefalia, hipotonia, defeito do nervo óptico, pé caído, malformação do aparelho cardiovascular e dificuldade para ganho de peso. O diagnóstico precoce da SWH é essencial para delinear um plano terapêutico estratégico e personalizado, alcançando melhor prognóstico e qualidade de vida do paciente e familiares. Neste caso, o plano terapêutico multiprofissional permitiu um seguimento assertivo e individualizado.
Palavras-chave: Anormalidades Congênitas. Transtornos Cromossômicos. Síndrome de Wolf-Hirschhorn. Relato de Casos.
Abstract
Wolf-Hirschhorn Syndrome (WHS) is caused by a genetic deletion of the short arm of chromosome 4 (4p16.3). It has an incidence of 1:50,000 live births and a 2:1 ratio between women and men. The extension of the chromosomal deletion determines the phenotypic variations, which may involve craniofacial abnormalities, epileptic seizures, delayed neuropsychomotor development, microcephaly, hypotonia and malformations of multiple systems, especially the cardiac. This study will describe a case of WHS in a female child, diagnosed using a DNA microarray genetic study. Among the manifestations of the genetic anomaly, the child presented a typical craniofacial sign, partial and generalized seizures, severe delay in neuropsychomotor development, microcephaly, hypotonia, optic nerve defect, foot drop, malformation of the cardiovascular system and difficulty in gaining weight. Early diagnosis of WHS is essential to outline a strategic and personalized therapeutic plan, achieving better prognoses and quality of life for patients and families. In this case, the multidisciplinary therapeutic plan allowed assertive and individualized follow-up.
Keywords: Congenital Abnormalities. Chromosome Disorders. Wolf-Hirschhorn Syndrome. Case Reports.
INTRODUÇÃO
A síndrome de Wolf-Hirschhorn (SWH) é uma condição rara de etiologia genética, classificada como uma doença de deleção cromossômica, que atinge o braço curto da região 4p16.3.1-4 Na maioria dos casos, a SWH é diagnosticada após o nascimento, em crianças com uma particularidade craniofacial, o "sinal da glabela" ou "sinal do capacete de guerreiro grego".1,3,4 A deleção cromossômica está associada a crises convulsivas parciais ou generalizadas, atraso grave do desenvolvimento neuropsicomotor, hipotonia, anomalias em múltiplos sistemas, principalmente cardíaco, e dificuldade para o ganho de peso.5,6
Considerando a complexidade da síndrome, com incidência de 1:50.000 nascidos vivos e prevalência de 2:1 entre mulheres e homens,1-4 o diagnóstico precoce e cuidado especializado são primordiais. A incidência pode ser ainda maior, salvo dificuldades para diagnóstico.7 A mortalidade é alta nos primeiros dois anos de vida (30% dos casos), associada a infecções do trato respiratório inferior, malformações cardíacas e anomalias congênitas múltiplas.7 Dada a singularidade, profissionais da saúde, nem sempre possuem domínio amplo acerca da conduta nessas condições.
DESCRIÇÃO DO CASO
Criança, sexo feminino, pequena para a idade gestacional, nascida prematuramente. Mãe, 27 anos, primigesta de uma gravidez planejada, sem comorbidades. Durante o primeiro trimestre da gestação, a criança apresentou crescimento normal pela ultrassonografia. A partir das 14 semanas de gestação, passou a apresentar retardo do crescimento intrauterino e centralização fetal. Às 33 semanas e 6 dias, a mãe teve um pico hipertensivo 180 x 100 mmHg, com indicação de interrupção da gestação por risco de sofrimento fetal.
A criança nasceu por cesariana pesando 840g, escore de APGAR de 6 e 9. Foi submetida imediatamente a manobras de reanimação neonatal, sendo realizada intubação orotraqueal (IOT) e internação em Unidade de Terapia Intensiva (UTI). Permaneceu em ventilação mecânica por sete dias após o nascimento. Recebeu nutrição enteral por sonda, sendo iniciado o aleitamento materno em seguida. Apresentou perda ponderal progressiva, sendo introduzida fórmula infantil para adequação nutricional, com ganho de peso satisfatório. Teve quadro séptico, tendo feito uso de antibioticoterapia (Oxacilina e Amicacina; Cefepime e Vancomicina; e Piperacilina e Tazobactam). Realizou ecocardiograma, evidenciando estenose de artéria pulmonar. A criança permaneceu em ambiente hospitalar por 80 dias no total. Desde o nascimento, foi observado sinal facial típico, caracterizado pela glabela proeminente, entre sobrancelhas arqueadas, contínua com uma ponte nasal ampla e olhos acentuadamente espaçados, com estrabismo, levantando-se a suspeita de síndrome genética.
A suspeita de síndrome genética foi avaliada aos sete meses através de análise genética pelo método de bandamento-G. Em todas as células analisadas, foi detectado um segmento cromossômico adicional no braço curto do cromossomo 4. Com 1 ano e 4 meses, realizou correção cirúrgica de cardiopatia congênita. Para dar seguimento à investigação citogenética, com 1 ano e 10 meses, realizou exame para sequenciamento de nova geração (NGS), em que foi detectada uma alteração de número de cópias em heterozigose na região cromossômica 4p16.3-16.1, concluindo-se que esta correspondia ao fenótipo apresentado pela criança. Aos 2 anos e 2 meses, realizou o exame de hibridização genômica por microarranjo, sendo identificada uma deleção de 13 megabases no cromossomo 4 na região p16.3p15.33, posição linear 68345-13159810 (Figura 1). Esta deleção é considerada patognomônica de síndrome de Wolf-Hirschhorn.
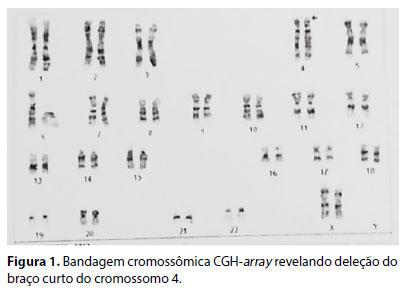
A criança realizava acompanhamento regular com médicos pediatra, oftalmologista, neurologista, cardiologista e geneticista. Evoluiu com atraso importante do desenvolvimento neuropsicomotor. Apresentou assimetria do eixo longitudinal da coluna toracolombar. Não apresentou alterações em pelve, sacro e cóccix. Apresentou pé equinovaro, com comprometimento da flexibilidade dos calcâneos. Desde os nove meses, realizava sessões semanais de fisioterapia e aos dois anos iniciou o uso de órteses em membros inferiores. Apresentou microdontia, realizando acompanhamento com dentista.
Aos 2 anos e 6 meses, a criança teve o primeiro episódio de crise convulsiva, de caráter tônico-clônica, autolimitada. Iniciou tratamento anticonvulsivante com ácido valproico e Fenobarbital, contínuos. No intervalo de um mês, a criança teve nova crise, permanecendo por 1h30min em estado de mal epiléptico (EME). Foi internada em UTI, diagnosticada com pneumonia e tratada com esquema antibiótico (Amoxicilina + Clavulanato). Após seis meses, teve nova crise tônico-clônica generalizada e novo diagnóstico de pneumonia, sendo internada novamente para realizar antibioticoterapia. Após dois meses da última internação, a criança, aos 3 anos e 4 meses, teve outra crise convulsiva, com manutenção do EME. Novamente necessitou internação para realizar tratamento de pneumonia recidivante. No total, ela teve quatro episódios de crise convulsiva em nove meses, sendo três concomitantemente a quadros de pneumonia.
Aos 3 anos e 4 meses, recebeu diagnóstico de hidrocefalia compensada, sendo observado foco occipital de hidrocefalia. Aos quatro anos, recebeu diagnóstico de autismo por médico neurologista pediátrico após análise comportamental. Nesse período, a criança iniciou em creche, sendo observada pobre interação com os colegas e professores. A criança tinha fala incompreensível, apenas emitindo gritos. Não sentava sem apoio, não engatinhava ou virava-se de bruços. Apresentava-se irritada e frustrada em ambientes distintos do familiar. Apresentou, ainda, seletividade alimentar extrema, restringindo-se a poucos alimentos, sempre na forma pastosa. Recusava-se a experimentar novas receitas e texturas, e recusava se a realizar as refeições na ausência dos pais.
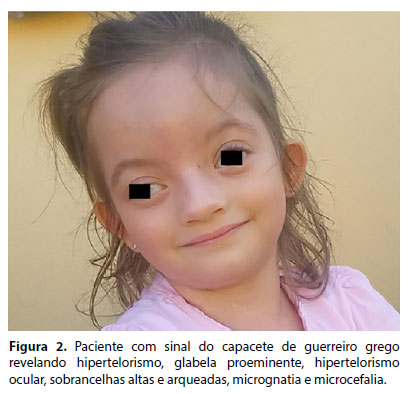
Atualmente, aos cinco anos (Figura 2), não teve recorrência das crises convulsivas. Encontra-se em atendimento multidisciplinar. Quanto ao desenvolvimento neuropsicomotor atual, a criança interage através de gritos e fala incompreensível, segue com os olhos, bate palmas, manda beijos, sorri e acena com as mãos. Senta sem apoio, mas se joga para trás eventualmente. Não fica em pé sozinha e não caminha. Encontra-se em uso contínuo de Clobazam, Canabidiol e Melatonina.
DISCUSSÃO
A SWH é uma doença genética rara caracterizada pela deleção do braço curto do cromossomo 4, ocasionando fenótipo facial típico, crises convulsivas, microcefalia, hipotonia generalizada, comprometimento do desenvolvimento neuropsicomotor, atraso no crescimento e baixo ganho de peso.1-6 A primeira descrição da deleção do cromossomo 4 ocorreu em 1961, por Hirschhorn e Cooper.7,8 Um segundo caso foi descrito por Bovin e Wolf em 1965.9 As manifestações clínicas dos casos foram comparadas, sendo observado em ambos os pacientes proeminência da glabela, hipertelorismo ocular, sobrancelhas altas e arqueadas, micrognatia e microcefalia. Este sinal facial típico foi denominado "capacete de guerreiro grego".1-6,8,10-12 Estes dois relatos de caso deram nome à síndrome.10,11 Foram descritos mais de 300 casos no mundo desde então.11,13
O desenvolvimento na técnica de bandagem cromossômica na década de 80 possibilitou associar o fenótipo da SWH com a deleção do cromossomo 4p16, mais especificamente, no ano de 2004, 4p16.3.11 Aproximadamente 55% dos pacientes diagnosticados apresentaram apenas esta alteração cromossômica,11 o que também foi observado na paciente deste estudo.
A primeira linha para confirmação diagnóstica da SWH é o exame de hibridização genômica por microarranjo,6,13,14 utilizado neste caso. Dentre as manifestações da anomalia genética, a criança apresentou o sinal craniofacial típico, crises convulsivas parciais e generalizadas, atraso grave do desenvolvimento neuropsicomotor, hipotonia, defeito do nervo óptico pé caído, malformação do aparelho cardiovascular e dificuldade para ganho de peso.
A investigação diagnóstica é iniciada essencialmente com base na suspeita clínica, contemplando sinais presentes logo ao nascer (sinal facial "capacete de guerreiro grego", hipotonia de membros inferiores, cardiopatia congênita e baixo peso). Casos de manifestações pré-natais são raros, com baixo desempenho ultrassonográfico.1 Neste caso, a criança apresentou restrição do crescimento intrauterino, presente em 80% dos casos.2
A epilepsia tem incidência em 90% dos pacientes até três anos de vida.2,3,6,11 A base genética das crises consiste na região crítica WHSCR1, WHSCR2 e o gene LETM1, que origina uma proteína necessária para homeostasia do cálcio, cuja deleção levaria a convulsões.4,5,6 Esta criança teve três internações em UTI por epilepsia de difícil controle. Encontra-se sem crises desde os quatro anos, atualmente em uso de Clobazam e Canabidiol. Estudos em pacientes com SWH demonstraram que as crises, apesar de serem reprimidas com certa dificuldade nos primeiros anos de vida, vêm a ser bem controladas, de forma que a expectativa de vida dos pacientes seja similar àqueles com distúrbios epilépticos.11
O atraso no desenvolvimento do sistema neuropsicomotor é outro achado comum na SWH.1-4,6,8 Defeitos estruturais do sistema nervoso central podem ser detectados através de exames de imagem em 80% dos pacientes. Estes achados não se relacionam com a presença de crises convulsivas.11 Neste caso, uma ressonância magnética de crânio demonstrou hipodesenvolvimento do corpo caloso, dilatação dos ventrículos cerebrais e ausência de hipertensão liquórica. A paciente apresentou diversas disfunções do sistema motor, como hipotrofia de membros inferiores, com grandes dificuldades para atingir os marcos de desenvolvimento, mesmo realizando sessões de fisioterapia semanais desde os nove meses de idade. A criança também apresentou atraso importante no desenvolvimento da fala, dificuldade de aprendizagem e problemas de interação social, de forma que realiza acompanhamento regular com neurologista e terapeuta ocupacional. Todos os indivíduos com SWH apresentam atraso global de desenvolvimento, com limitação da linguagem a sons monossilábicos. A compreensão está limitada a ordens simples.10
A hipotonia generalizada é um achado comum,1-6 geralmente associada a hipotrofia muscular.1,2,3 Existem relatos de crianças com SWH que apresentaram displasia de quadril,11,14 o que não foi observado na paciente. A criança apresentou pé equinovaro, condição associada à dificuldade de caminhar e presente em 70% dos casos.4,8 Alterações na coluna vertebral, como cifose e escoliose, também foram descritas.4 Neste caso, a criança apresentou assimetria do eixo longitudinal da coluna toracolombar com convexidade à esquerda.
A criança apresentou defeito do nervo óptico, o que ocorre em média de 40% dos casos.1,5 Apresentou, ainda, estrabismo, comum na SWH.10,11 Cerca de um terço dos pacientes apresenta disfunções no trato urinário,1,2 o que não foi observado nesta criança. Também não foram observadas alterações do sistema gastrointestinal. A paciente teve quadros frequentes de otite média aguda, muito comum em pacientes SWH.10 No presente estudo, observaram-se associação de infecção de vias aéreas e febre baixa como fator desencadeante de crises convulsivas e maior susceptibilidade a infecções, ambos relatados em literatura.2,4
Defeitos cardíacos congênitos ocorrem em média em 50% dos casos,2-6,11,15 sendo defeitos septais leves e persistência do ducto arterioso as patologias mais comuns.2,12,14,15 O gene WHSC1 é o mais provável associado às cardiopatias, por sua interação com o fator de transcrição cardíaca Nkx2.5.15 Outro gene provável é o FGFRL1, que codifica um dos receptores de crescimento fibroblástico no coração.15 Neste caso, observou-se alteração cardíaca congênita da válvula pulmonar, sendo a segunda anormalidade cardíaca mais comum entre pacientes com a síndrome.1,2
Apesar da adequação nutricional, pacientes com SWH apresentam baixa estatura e ganho lento de peso.1,3,11 Alguns sinais que justificam tal achado são fissuras na estrutura orofacial, dificuldade para sugar e engolir, com consequente aspiração do alimento, e refluxo esofágico.1,5,11 Nenhum destes foi observado no presente estudo. Em contrapartida, a criança apresenta microdontia, sendo que 50% dos indivíduos com SWH apresenta alguma alteração dentária.1,2,5,6,10,11 Alterações dermatológicas são relativamente comuns,8,14 o que não foi observado nesta paciente.
Uma vez que a SWH é genética e não tem cura, o tratamento é basicamente sintomático e multidisciplinar.6 Cerca de 35% dos pacientes SWH falecem no primeiro ano de vida, porcentagem associada ao abandono do tratamento.4 Há registros de pacientes SWH que chegaram aos 60 anos.6 Atualmente, aos cinco anos, a paciente apresenta bom estado geral e bom prognóstico, sem recidiva das crises convulsivas e sem novas internações. Apresenta boa adesão ao plano de tratamento multidisciplinar e medicamentoso.
REFERÊNCIAS
1. Tang F, Zeng Y, Wang L, Yin D, Chen L, Xie D, et al. Prenatal phenotype of Wolf-Hirschhorn syndrome: A case series and literature review. Molecular Genetics & Genomic Medicine [Internet]. 2023 [cited 2024 Feb 10];11(6):1-9. DOI https://doi.org/10.1002/mgg3.2155. Disponível em: https://onlinelibrary.wiley.com/doi/10.1002/mgg3.2155
2. Gavril EC, Luca AC, Curpan AS, Popescu R, Resmerita I, Panzaru MC, et al. Wolf-Hirschhorn Syndrome: Clinical and Genetic Study of 7 New Cases, and Mini Review. Children (Basel) [Internet]. 2021 [cited 2024 Feb 10];8(9) DOI https://doi.org/10.3390/children8090751. Disponível em: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC8471045/.
3. Chaudhry C, Kaur A, Panigrahi I Kaur A. Wolf-Hirschhorn syndrome: A case series from India. American journal of medic genetics. Part A [Internet]. 2020 [cited 2024 Feb 10];182(12). DOI https://doi.org/10.1002/ajmg.a.61856. Disponível em: https://pubmed.ncbi.nlm.nih.gov/32914558/.
4. Corrêa T, Riegel M. Análise Citogenômica Integrativa da Região Crítica 4p16.3 associada a Síndrome de Wolf Hirschhorn [Dissertação]. Programa de Pós-Graduação em Genética e Biologia Molecular (UFRGS); 2021 [cited 2024 Feb 10]. 146 p. DOI http://hdl.handle.net/10183/180605. Disponível em: https://lume.ufrgs.br/handle/10183/180605
5. Rutherford EL, Lowery LA. Exploring the developmental mechanisms underlying Wolf-Hirschhorn Syndrome: Evidence for defects in neural crest cell migration. Developmental Biology [Internet]. 2016 [cited 2024 Feb 10];420(11) DOI https://doi.org/10.1016/j.ydbio.2016.10.012. Disponível em: https://pubmed.ncbi.nlm.nih.gov/27777068/.
6. Lago RB, Mori XS, Fenellos CB, Malaga DI, Alvarez MAM, Barreiro NG, et al. Prevalence and geographic distribution of the Wolf-Hirschhorn syndrome in Spain. Revista Espanola de Salud Publica [Internet]. 2021 [cited 2024 Feb 12];8(96) DOI 35703131. Disponível em: https://www.sanidad.gob.es/biblioPublic/publicaciones/recursos_propios/resp/revista_cdrom/VOL96/ ORIGINALES/RS96C_202206045.pdf
7. Hirschhorn K, Cooper HL, Firschein IL. Deletion of short arms of chromosome 4-5 in a child with defects of midline fusion. Humangenetk [Internet]. 1961 [cited 2024 Feb 12];1(5):479- 482. DOI https://doi.org/10.1007/bf00279124. Disponível em: https://pubmed.ncbi.nlm.nih.gov/5895684/.
8. Battaglia A, Calhoun ARUL, Lortz A, Carey JC. Risk of hepatic neoplasms in Wolf-Hirschhorn syndrome (4p-): Four new cases and review of the literature. American journal of medic genetics. Part A [Internet]. 2018 [cited 2024 Feb 12];176(11):2389-2394. DOI https://pubmed.ncbi.nlm.nih.gov/30289612/. Disponível em: https://pubmed.ncbi.nlm.nih.gov/30289612/.
9. Wolf U, Reinwein H, Porsch R, Schröter R, Baitsch H. Deficiency on the short arms of a chromosome No. 4. Humangenetik [Internet]. 1961 [cited 2024 Feb 12];1(5):397-413. DOI 5868696. Disponível em: https://pubmed.ncbi.nlm.nih.gov/5868696/.
10. Stolarz AMP. Wolf-Hirschhorn syndrome (WHS) - literature review on the features of the syndrome. Advances in Clinical and Experimental Medicine [Internet]. 2014 [cited 2024 Feb 13];23(3) DOI https://doi.org/10.17219/acem/24111. Disponível em: https://pubmed.ncbi.nlm.nih.gov/24979523/.
11. Battaglia A, Carey JC, South ST. Wolf-Hirschhorn syndrome: A review and update. American journal of medic genetics. Part C, Seminars in Medical Genetics [Internet]. 2015 [cited 2024 Feb 13];169(3):216-223. DOI https://doi.org/10.1002/ajmg.c.31449. Disponível em: https://pubmed.ncbi.nlm.nih.gov/26239400/.
12. Duarte RCB, Borges PVG, Novaes RS, Carvalho RM, Colonnelli G, D'Oliveira MS. Wolf-Hirschhorn syndrome (terminal deletion of the short arm of chromosome 4p): Case report. Revista Paraense de Medicina [Internet]. 2007 [cited 2024 Feb 13];21(3):53-57. DOI 0101-5907. Disponível em: http://scielo.iec.gov.br/scielo.php?script=sci_arttext&pid=S0101-59072007000300009
13. Miller DT, Adam MP, Aradhya S, Biesecker LG, Brothman AR, CNP, et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. American Journal of Human Genetics [Internet]. 2010 [cited 2024 Feb 13];86(5) DOI 10.1016/j.ajhg.2010.04.006. Disponível em: https://pubmed.ncbi.nlm.nih.gov/20466091/.
14. Dorfman LM, Leite JCL, Giugliani R, Riegel M. Microarray‐based comparative genomic hybridization analysis in neonates with congenital anomalies: detection of chromosomal imbalances. Jornal de Pediatria [Internet]. 2015 Feb 01 [cited 2024 Feb 13];91(1):59-67. DOI https://www.jped.com.br/pt-microarraybased-comparative-genomic-hybridization-analysis-articulo-S2255553614001542. Disponível em: https://www.jped.com.br/pt-microarraybased comparative-genomic-hybridization-analysis-articulo-S2255553614001542?referer=buscador
15. Saliba A, Figueiredo ACV, Baroneza JE, Afiune JY, Pic‐Taylor A, Oliveira SF, et al. Genetic and genomics in congenital heart disease: a clinical review. Jornal de Pediatria [Internet]. 2020 [cited 2024 Feb 13];96(3):279-288. DOI https://www.jped.com.br/pt-genetic-genomics-in-congenital-heart-articulo-S2255553619301466. Disponível em: https://www.jped.com.br/pt-genetic-genomics-in-congenital-heart-articulo S2255553619301466?referer=buscador
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()