Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 24(1) - Março 2024
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Síndrome de Rett atípica: diagnóstico diferencial em pacientes pediátricas com características "autista-like"
Atypical Rett Syndrome: Differential Diagnosis in Pediatric Patients with "Autistic-like" Features
Leticia dos Santos Pagnano1; Julia Sampaio1; Tamires Ribeiro de Paula Vilela1; Zumira Aparecida Carneiro1; Charles Marques Lourenco1,2
DOI:10.31365/issn.2595-1769.v24i1p22-28
1. Centro Universitário Estácio de Ribeirão Preto, Faculdade de Medicina - RIBEIRAO PRETO - SP - Brasil
2. Faculdade Estadual de Medicina de São José do Rio Preto (FAMERP), Neurogenética - São José do Rio Preto - SP - Brasil
Endereço para correspondência:
Charles Marques Lourenco
charlesgenetica@gmail.com
Instituição: Centro Universitário Estácio de Ribeirão Preto
Recebido em: 26/11/2020?
Aprovado em: 22/01/2024
Resumo
INTRODUÇÃO: Síndrome de Rett (RTT) é um distúrbio do neurodesenvolvimento causado por mutações no gene MECP2, acometendo predominantemente pacientes do sexo feminino. Pode-se dividir em dois subgrupos de pacientes: aqueles que manifestam sinais e sintomas característicos de RTT e outro que apresenta sintomas "não clássicos". Caso 1: Paciente, sexo feminino, 17 anos, encaminhada aos 7 anos por apresentar quadro de convulsões, hipotonia e hipoatividade, Apresentava-se pouco responsiva a estímulos até os 7 meses de idade e, mesmo após esse período, ainda era hipoativa em comparação ao neurodesenvolvimento. Investigação por sequenciamento completo de exoma (SEC) evidenciou mutação patogênica em heterozigose no MECP2. Caso 2: Paciente, sexo feminino, 6 anos e 11 meses, encaminhada para avaliação de atraso neuropsicomotor e característica autista-like. Exames de ressonância magnética de encéfalo, cariótipo, SNP-array e de erros inatos do metabolismo dentro da normalidade. Diante da não elucidação etiológica do seu quadro clínico, optou-se pela realização do SEC, sendo detectada a mutação no gene MECP2.
DISCUSSÃO: Pacientes com RTT possuem desenvolvimento neurológico normal nos primeiros meses de vida, seguindo-se por característica regressão das habilidades motoras e de comunicação entre 6 e 18 meses de idade, podendo ser acompanhada por hipotonia, crises convulsivas e elementos do espectro autista. Apesar de ambas as pacientes relatadas serem consideradas como formas atípicas, no caso 1 o espectro clínico é mais grave desde a primeira infância, com hipotonia congênita e atraso do neurodesenvolvimento, enquanto na paciente 2 a forma clínica é mais atenuada, com menor comprometimento intelectual, de comunicação e da regressão neuropsicomotora.
Palavras-chave: Síndrome de Rett. Epilepsia. Transtorno do Espectro Autista. Proteína 2 de Ligação a Metil-CpG
Abstract
INTRODUTION: Rett syndrome (RTT) is a neurodevelopment disturbance caused by mutations in the MECP2 gene, affecting mostly female patients. The patients can be distributed in two main subgroups; classic RTT form, in which the progression presents characteristic and expected symptoms, and variant (or atypical) RTT form, evolving with unexpected symptoms. This report is a comparative study of two atypical RTT cases.
CASE REPORT: Patient #1: Female, 17 years old, presented with seizures, hypotonia, and hypoactivity at the age of 7 years. It was reported a lack of responsiveness to stimuli until 7 months old. Even after this age, the hypotonia was notable in comparison to her neurodevelopment, contributing to a delay in the investigation of a pathogenic mutation in the MECP2 gene. Patient #2: Female, 7 years old, with neuropsychomotor delay and autistic characteristics. Brain MRI, karyotype, SNP-array, and innate errors of metabolism evaluations were unremarkable. Due to the non-elucidation of the clinical condition, whole-exome sequencing was performed, and the MEC2 gene mutation was detected.
DISCUSSION: RTT is characterized by motor and communication skills regression in patients with normal neurological development in the first few months of living, affecting patients 6 to 18 months old, alongside possible hypotonia, seizures, and autistic spectrum elements. Even though both patients are considered as RTT atypical forms, the clinical condition of the first case is more severe since early childhood, with congenital hypotonia and neurodevelopment delay, while the clinical condition of the second patient is milder, with less intellectual and communication compromising and neuro psychomotor regression.
Keywords: Rett Syndrome. Epilepsy. Autism Spectrum Disorder. Methyl-CpG-Binding Protein 2.
1. INTRODUÇÃO
Descrita pela primeira vez por Andreas Rett em 1966,1 a síndrome de Rett (RTT) (OMIM # 312750) consiste em um distúrbio do neurodesenvolvimento ligado ao cromossomo X que afeta predominantemente as mulheres.2 O diagnóstico da referida síndrome, outrora, era essencialmente clínico e a gravidade clínica da doença estava fortemente relacionada ao diferente espectro fenotípico de manifestações neurológicas.3
Apenas no ano de 1999, foram identificadas mutações no gene MECP2, permitindo agregar o teste genético como imperiosa ferramenta na confirmação diagnóstica de RTT.3 Mutações no MECP2 foram encontradas na maior parte dos pacientes que compartilhavam sintomas e evolução clínica considerados como típicos para a síndrome de Rett, também chamados de "RTT clássicos".3,4
Frisa-se que o gene MECP2 se localiza no cromossomo humano q28, apesar de o exato mecanismo das mutações que originam o RTT não ser completamente conhecido.4 Nesse contexto, estudos recentes apontam que o gene MECP2 exerce papel fundamental, como regulador do processo de transcrição proteica neuronal.3,5 Sendo assim, mutações que promovessem a alteração da função do referido gene levariam a um processo de disfunção neuronal, com progressiva perda celular, resultando nos déficits cognitivos e motores observados nos pacientes afetados por essa enfermidade,3 como é possível vislumbrar na Figura 1, a seguir.
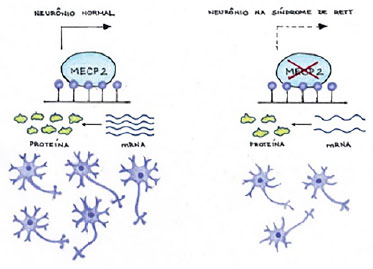
Figura 1. (A) Mecanismo do gene MECP2 em um neurônio normal; (B) Quando ocorre mutação do gene MECP2 induz a alteração do RNA mensageiro levando a diminuição das conexões sinápticas. Fonte: Acervo dos autores (2020).
O curso da síndrome decorre, aparentemente, do tipo/gravidade de mutação no MECP2, embora a correlação genótipo-fenótipo raramente possa ser feita.3,5 A forma típica se inicia com estagnação precoce do neurodesenvolvimento, habitualmente, ocorrendo a partir do sexto mês de vida.1 Como bebês afetados nessa fase apresentam-se extremamente calmos, raramente profissionais da saúde ou até mesmo seus próprios genitores reconhecem alguma anormalidade aparente.3,4 Progressivamente, contudo, os pacientes evoluem para um estágio de perda gradual de habilidades previamente adquiridas. Releva-se que esse processo ocorre de maneira e velocidade diferente em cada um dos pacientes.3,6 Ademais, tais pacientes desenvolvem corriqueiramente deterioração motora, ataxia, apraxia, hipotonia muscular com posterior evolução para espasticidade, comprometimento da fala e da comunicação, bem como o surgimento de crises convulsivas, muitas vezes de moroso manejo clínico.7
Na forma "clássica" de RTT, os mais diversos tipos de terapias de reabilitação podem ser adotados, tais como: a fonoaudiologia, terapia ocupacional, fisioterapia e psicoterapia. Entretanto, mesmo que atuem proativamente no manejo terapêutico dos pacientes, não se mostram capazes de impedir a progressão da doença.2,8
Deste modo, como critérios para o diagnóstico de RTT clássica, destacam-se as seguintes manifestações: presença de estereotipias manuais, tidas como movimentos repetitivos das mãos, perda parcial ou completa do uso funcional das mãos, regressão da linguagem e, por derradeiro, alteração de marcha.3
Comparativamente, a RTT atípica é diagnosticada quando dois desses quatro critérios típicos estão presentes, atrelados a cinco dos 11 critérios de suporte, que são, respectivamente: distúrbios respiratórios, deglutição do ar, bruxismo, escoliose, marcha anormal, amiotrofia dos membros inferiores, pés frios e cianóticos, resposta diminuída à dor, distúrbios do sono, risadas e gritos inapropriados e contato visual intenso (Tabela 1).9
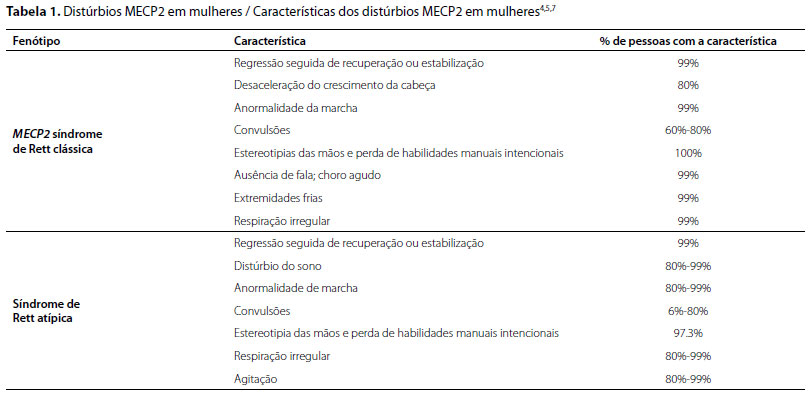
No sexo feminino, o espectro dos fenótipos relacionados ao MECP2 varia da síndrome de Rett clássica à síndrome de Rett variante (que pode se manifestar de forma mais branda ou mais grave que a síndrome de Rett clássica), incluindo pacientes que apresentam apenas dificuldade leve de aprendizado.10 Enquanto isso, no sexo masculino, o espectro varia de encefalopatia neonatal grave à síndrome de sinais piramidais, parkinsonismo e síndrome de macro-orquidismo (PPM-X), abrangendo diversos "níveis" de comprometimento cognitivo.10
É possível aduzir que mulheres portadoras da síndrome de Rett variante exibem um espectro mais amplo de características clínicas do que aquelas observadas na síndrome de Rett clássica.10 Acrescenta-se que, no espectro mais grave, ocorrem atraso no desenvolvimento desde a primeira infância, hipotonia congênita e espasmos infantis.10 Na outra ponta do espectro, a regressão é menos dramática e a incapacidade intelectual é mais moderada, além de ocorrer preservação parcial da fala.4
Em casos mais raros, mulheres com uma variante patogênica do MECP2 podem demonstrar apenas dificuldades leves de aprendizado ou algumas características autísticas (possivelmente como consequência do fenômeno de inativação aleatória do cromossomo X).10 Ressalta-se que o fenótipo mais atenuado diverge do curso clínico da síndrome de Rett clássica e da variante, diante do fato de as pacientes não apresentarem fase de regressão neurológica, tampouco as estereotipias manuais características da doença.6,10
Isto posto, o objetivo do presente estudo é relatar pacientes portadoras da mutação no gene MECP2 e que exibem tanto o fenótipo atenuado quanto o de início precoce; ressaltar as diferentes formas de apresentação da síndrome de Rett (fenótipo típico e atípico) e, por fim, demonstrar o desafio encontrado entre os profissionais envolvidos no reconhecimento dos sinais e sintomas da doença.
2. DESCRIÇÃO DOS CASOS
Paciente 1
A primeira paciente é do sexo feminino, atualmente com 17 anos de idade e filha de pais não consanguíneos. Preliminarmente, a paciente foi encaminhada para avaliação de hipotonia e atraso de marcos motores.
No que diz respeito aos anos iniciais da paciente, evidencia-se que a mesma nasceu de parto cesárea, com 39 semanas, pesando aproximadamente 3.195 gramas (p50) e com 49 cm de comprimento (entre os percentis 75 e 90). Destarte, foi relatado o choro e evoluiu sem outros agravos à saúde. A paciente obteve alta com 3 dias de vida, mas a genitora relatou que a criança já apresentava sinais de hipotonia desde o início. Negou, entretanto, a ocorrência de outros problemas de saúde nos períodos neo- e perinatal.
A paciente demonstrou-se pouco responsiva a estímulos até os sete meses de idade e, mesmo após esse período, ainda permanecia hipoativa, se comparada ao neurodesenvolvimento padrão. O sono, por exemplo, sempre foi marcado por movimentos estereotipados da cabeça de flexão e extensão. Acrescenta-se que a criança se sentou sem apoio aos oito meses, aos dois anos andou com apoio e aos três anos emitiu sons guturais (sons incompreensíveis), sem o desenvolvimento da fala.
Aos sete meses de idade, foram observadas em consulta com a pediatra responsável: hipotonia e hipoatividade. Diante da situação, a pediatra encaminhou a paciente para acompanhamento com fisioterapeuta. Nessa faixa etária, observou-se o início de movimentos involuntários dos olhos da paciente.
Por conseguinte, dos sete meses aos cinco anos, o único marco de desenvolvimento perceptível foi o de rolar na cama sem auxílio (primeiro movimento da paciente em que não foi necessário apoio). Aos cinco anos de idade, a criança passou por sua primeira crise epiléptica, motivo pelo qual perdeu a habilidade de deambular com suporte unilateral.
Progressivamente, houve piora das crises convulsivas, a despeito do uso de vários anticonvulsivantes (Tabela 1), culminando na internação em Unidade de Tratamento Intensivo (UTI) para controlar a encefalopatia epiléptica. Posteriormente, aos sete anos de idade, por ainda não apresentar diagnóstico concreto de sua enfermidade, foi encaminhada para serviço de neurogenética para elucidação diagnóstica.
Dos 7 aos 13 anos de idade, a paciente apresentou uma piora, marcada pelo aumento na frequência das crises epilépticas. As crises epilépticas alternavam-se entre crises focais disperceptivas e crises generalizadas tônico clônicas. Durante esse período, houve a transição de medicação, cessando o uso do Ácido Valproico, devido a complicações depressoras do sistema nervoso central associadas ao uso da droga, tal como nefrotoxicidade e intolerância gástrica (vômitos recorrentes).
Nesse cenário, a paciente passou a utilizar Fenobarbital e foi orientada pela médica neurologista que acompanhava o caso a fazer o uso de O2 e Fenobarbital em dose adicional em eminência de crise. Ulteriormente, dos 13 aos 17 anos de idade, houve melhora acentuada na frequência das crises, que ocorriam com intervalo de 15 a 20 dias.
Durante esse período, a paciente também apresentou uma piora no quadro da escoliose. Reforça-se que, aos 17 anos, a escoliose da paciente apresentava-se em 94°, sendo necessário o uso de cadeira adaptada. A expressiva piora na curvatura da coluna ocasionou compressão torácica e, consequentemente, atelectasia pulmonar, resultando em uma queda da saturação, sobretudo durante o sono.
Com a queda da saturação, recomendou-se o uso de O2 0,5L/min, a ser ministrado por intermédio de cateter nasal, durante o período noturno. Além disso, no período supracitado, a paciente teve de ser internada duas vezes, em decorrência de pneumonias bacterianas graves, mas não foi necessário o uso de intubação orotraqueal e outras medidas invasivas.
O exame de exoma foi realizado em 11 de maio de 2017, quando a paciente se encontrava com 14 anos de idade. Inicialmente, foram investigados os genes relacionados com a epilepsia de início precoce, associada à hipotonia congênita, como os genes FOXG1 e SCN1A. Os resultados revelaram-se dentro da normalidade. Sendo assim, foi requisitado o exame de sequenciamento completo de exoma e, por meio de seu resultado, foi possível vislumbrar a presença de mutação patogênica em heterozigose no MECP2: c.538C>T, (p.Arg180*).
A presença desta variante foi confirmada por metodologia complementar, com o sequenciamento Sanger, e não foi identificada em seus genitores. A seguir, na Tabela 2, é possível visualizar os anticonvulsivantes utilizados pela paciente:
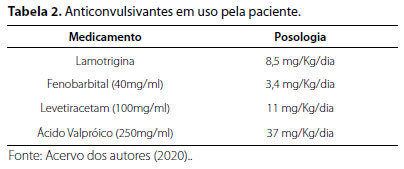
Hodiernamente, aos 17 anos, a paciente apresenta crises a cada 10 a 15 dias, necessitando, corriqueiramente, de administração de dose extra de Fenobarbital quando apresenta crise prolongada. Neste caso, é usualmente encaminhada para Unidade de Emergência para monitoração clínica. Além das crises recorrentes, a genitora descreve um comportamento de autoagressividade, em que a paciente morde suas próprias mãos constantemente, do mesmo modo que apresenta quadro "depressivo" (sic), com aparente prostração e diminuição da interação social.
Diante do quadro clínico, a paciente faz acompanhamento com equipe multidisciplinar, realizando as seguintes atividades: reabilitação com fisioterapia, terapia ocupacional e atendimento psicológico. É imperioso asseverar que a adolescente não apresenta linguagem verbal e que sua principal forma de interação é manifesta por meio de movimentos oculares e com a visualização de televisão.
Em síntese, seu exame genético-clínico revelou estatura de 128 cm (p25); peso de 25kg (~p25); perímetro cefálico 49,2 cm (<p3); ausência de dismorfias faciais; presença de estrabismo divergente; hiperreflexia generalizada; postura distônica de pés e mãos; tetraespasticidade; força muscular preservada; restrita a cadeira de rodas e pouca interação com o examinador.
Paciente 2
A segunda paciente investigada no presente estudo é do sexo feminino, idade de seis anos e 11 meses (aproximadamente sete anos), primogênita de pais não consanguíneos e foi encaminhada para avaliação de atraso neuropsicomotor e características autista-like.
Assim como no primeiro estudo de caso, a presente paciente nasceu de parto cesariana com 39 semanas e sem intercorrências durante o pré-natal e parto. A paciente chorou ao nascer e foram apuradas as seguintes características: Apgar avaliado em 9/10; peso de 3.020 gramas (entre os percentis 25 e 50); comprimento 49 centímetros (entre os percentis 25 e 50) e perímetro cefálico 34 centímetros (~p10). A genitora e a criança receberam alta dois dias após o parto. A paciente apresentou quadro de icterícia sem complicações e, por esse motivo, foi necessária a adoção de fototerapia até os dois meses de idade.
Até os seis meses de vida, os genitores não observaram anormalidades manifestas; no entanto, no decorrer do tempo, a paciente passou a apresentar maior irritabilidade, choros frequentes e intensos sem causa aparente. Os episódios marcados pelo choro intenso cessaram espontaneamente quando a paciente chegou aos 12 meses de vida. Entretanto, na mesma faixa etária, a criança passou a apresentar sinais de hipotonia.
No tocante aos marcos do neurodesenvolvimento, observa-se que a paciente andou com aproximadamente um ano e sete meses e o início da fala ocorreu com um ano e quatro meses, embora tenha passado a elaborar frases com apenas quatro anos de idade. Inobstante o desenvolvimento da marcha, a paciente sempre deambulou, apresentando quedas frequentes.
Esta foi a razão pela qual os genitores se propuseram a levá-la para iniciar a investigação de seu quadro clínico. Após minuciosa investigação, a paciente foi diagnosticada com "transtorno do espectro autista (TEA), transtorno de oposição desafiante (TOD) e transtorno de déficit de atenção com hiperatividade (TDAH)" (sic).
Após a construção do diagnóstico, a criança iniciou a terapia de reabilitação com a equipe multiprofissional aos três anos de idade (fisioterapia, fonoaudiologia, psicopedagogia, psicologia, terapia ocupacional e musicoterapia). Com a terapia de reabilitação, diversos benefícios clínicos foram percebidos, como a melhora motora e comportamental, mesmo com as dificuldades relacionadas à fala (dislalia e atraso na linguagem) e movimentos estereotipados das mãos.
Aos cinco anos de idade, a paciente passou a desenvolver crises de ausência, confirmadas com o auxílio de eletroencefalograma (EEG). Com o desenvolvimento das crises, foi fundamental iniciar o uso de Lamotrigina (9,9 mg/kg/dia). Por apresentar um sono "bastante agitado", a criança passou a utilizar Melatonina (3mg/dia) durante o período noturno, o que propiciou melhora significativa no padrão de sono.
A genitora relatou que a criança apresenta alta dificuldade de deglutição e mastigação - por exemplo, possui diversos engasgos, inclusive com líquidos. Contudo, a paciente não fora submetida ao exame de videoglutograma para confirmação da disfagia. Outrossim, o exame de ressonância magnética de encéfalo, cariótipo, SNP-array e testes bioquímicos para averiguação de erros inatos do metabolismo revelaram-se dentro da normalidade.
Perante a não elucidação etiológica do seu quadro clínico, os profissionais envolvidos optaram pela realização do sequenciamento complexo do exoma. Seguidamente à sua realização, foi detectada a mutação c.1173dup p. (VAal392Argfs*) no gene MECP2, confirmando o diagnóstico de síndrome de Rett.
O exame genético-clínico da paciente revelou: estatura 120 cm (entre p25 e p50); peso 20,2kg (entre p10 e p25); perímetro cefálico 54cm (entre p3 e p50); ausência de dismorfias faciais; motricidade ocular dentro do normal; presença de estereotipias manuais; dismetria e disdiacocinesia; reflexos osteotendíneos exaltados; tônus preservado; força muscular grau V; marcha com base alargada e boa interação com o examinador.
3. DISCUSSÃO
De antemão, destaca-se que a síndrome de Rett é causada por uma mutação no gene metil CpG da proteína 2 (methyl-CpG-binding protein 2) (MECP2), localizado no cromossomo Xq28.11 O MECP2 desempenha papel importante no desenvolvimento neural, por possuir a capacidade de se ligar ao DNA metilado, organizar o controle epigenético da transição e, assim, estruturar a cromatina das células neuronais.11
As mutações no MECP2 são encontradas em aproximadamente 95% dos casos de Rett clássica e em cerca de 75% dos casos atípicos. Neste prisma, as meninas portadoras da síndrome de Rett atípica apresentam um espectro clínico mais amplo do que aquelas que possuem a Rett clássica.10,11
No espectro clínico mais grave de apresentação de Rett, o paciente pode apresentar hipotonia congênita e espasmos, com atraso no desenvolvimento desde a primeira infância (conforme observado no quadro da primeira paciente); enquanto isso, na norma atenuada, o paciente possui menor comprometimento na capacidade intelectual e na regressão do neurodesenvolvimento. Nestes casos, é possível observar até mesmo o desenvolvimento da fala, como observado na paciente 2.
A síndrome de Rett foi descrita como um desenvolvimento aparentemente normal das crianças, acompanhada de uma regressão nas habilidades motoras e de comunicação, que ocorre entre 6 e 18 meses de idade. Habitualmente, as crianças portadoras da síndrome são hipotônicas e facilmente irritáveis.
Nos casos descritos em linhas pretéritas, as pacientes afetadas apresentam características clínicas distintas no aspecto de irritabilidade, haja vista que, no primeiro caso, verifica-se um bebê calmo e com choros esporádicos. Já no segundo caso, a paciente demonstra extrema irritabilidade desde os 6 meses de vida.12
Apesar de o aspecto de irritabilidade se distinguir entre os dois casos, releva-se que a hipotonia se destaca em ambos. Isto posto, a hipotonia representa um fator alarmante, pois, é acompanhada de um evidente atraso motor, bem como de crises convulsivas, como no caso da primeira paciente. Já no caso subsequente, a hipotonia mantém ligação com o TEA, transtorno que não se apresenta com tamanho retrocesso motor e intelectual.12
Parcela relativamente grande (cerca de 20% a 50%) dos pacientes portadores da síndrome de Rett possuem características autistas graves, que prevalecem no período de regressão. Essas características se relacionam com déficits de interação social e de comunicação. Em alguns casos, pessoas diagnosticadas com a síndrome são, no início, diagnosticadas com o transtorno do espectro autista (TEA), principalmente aquelas menos afetadas, como no relato da segunda paciente.12,13
Posto isto, depreende-se que os sinais do TEA são mais comuns na variante atípica branda síndrome de Rett. Além do autismo, a epilepsia é encontrada entre os portadores da síndrome de Rett, afetando entre 60% a 80% das meninas entre 2-5 anos de idade. Nos presentes estudos de caso, a epilepsia é uma das maiores preocupações clínicas da síndrome.14
As crises epilépticas ocorrem com maior frequência na faixa etária compreendida entre 7-12 anos de idade, conquanto a primeira paciente ainda apresenta crises, mesmo aos 17 anos de idade.14 Logo, a presença das crises epilépticas mantém forte relação com o mal prognóstico, no que concerne, a habilidade do uso das mãos, deambulação e comunicação, na medida em que pode acarretar sequelas neurológicas.
As sequelas neurológicas mencionadas, podem ser observadas durante o relato da paciente 1, que apresentou a perda da habilidade de deambular após uma crise epiléptica ocorrida aos cinco anos de idade. Já a paciente 2 apresentou apenas o laudo de paroxismos epileptiformes ao eletroencefalograma aos três anos de idade e mantém crises focais disperceptivas e eventualmente crises generalizadas tônico-clônicas.
Mesmo que a paciente 1 tenha demonstrado alterações da movimentação ocular, é uníssono que as referidas alterações não são frequentes nos portadores da síndrome de Rett. Ocorre que muitas crianças com RTT utilizam a movimentação ocular como forma de comunicação e linguagem (eye pointing).15,16
Notadamente, não existem tratamentos farmacológicos específicos para pacientes portadores da síndrome de Rett, mas existem medicações que atuam no controle sintomático do quadro, controlando as crises epilépticas, transtornos de humor e comorbidades associadas. Não há um antiepiléptico específico para o RTT, haja vista que o tratamento farmacológico da epilepsia deve levar em consideração o tipo de crise convulsiva que o paciente apresenta.
Mediante a análise de ambos os casos, notou-se que ambas as pacientes se valeram da utilização da Lamotrigina para o controle das crises epilépticas. Outros antiepilépticos, como Lacosamida, Levetiracetam e Fenobarbital, vêm sendo adotados para o tratamento de crises convulsivas em pacientes com RTT.11
É evidente que as diversas comorbidades associadas aos pacientes pressupõem a necessidade de atendimento multidisciplinar. Diante de todo o exposto, denota-se que a síndrome de Rett pode se apresentar de diferentes formas clínicas. Assim, o pediatra e o neuropediatra devem estar atentos não só à apresentação clássica da doença, como também a suas formas atípicas, pois as características podem estar presentes desde o nascimento.
REFERÊNCIAS
1. Pazeto TCB, Hara ACP, Barrozo AF, De Oliveira J, Khoury LP, et al. Síndrome de Rett : artigo de revisão. Rett syndrome. 2013; 13.
2. Chou MY, Changa NW, Chen C, Lee WT, Hsin YJ, Siu LL, et al. The effectiveness of music therapy for individuals with Rett syndrome and their families. J Formos Med Assoc. 2019; 188: 1633-1643.
3. Schwartzman JS. Síndrome de Rett atualização. Revista brasileira de Psiquiatria. 2003; 25: 110-113.
4. Roux JC, Ehinger Y, Matagne V, Villard L. Rett syndrome from bench to bedside: Recent advances. F1000Research. 2018; 7: 398.
5. Ip JPK, Mellios N, Sur M. Rett syndrome: insights into genetic, molecular and circuit mechanisms. Nature reviews Neuroscience. 2018; 9: 368-382.
6. Einspieler C, Marschik PB. Regression in Rett syndrome: Developmental pathways to its onset. Neurosci Biobehav Rev. 2019; 98: 320-32.
7. Pini G, Bigoni S, Congiu L, Romanelli AM, Scusa MF, Di Marco P, et al. Rett syndrome: a wide clinical and autonomic picture. Orphanet J Rare Dis. 2016; 11(1): 1-16.
8. Lewis J, Wilson D. Caminhos para a aprendizagem na síndrome de Rett. Memnom Edições Científicas [Livro].São Paulo. 1999. 1ed.
9. Smeets EEJ, Pelc K, Dan B. Rett Syndrome. Molecular Syndromology. 2011; 2: 113-127.
10. Kaur S, Christodoulou J. MECP2 Disorders. GeneReviews. 2001; 1-21.
11. Titlestad KB, Eldevik S. Modest but Clinically Meaningful Efects of Early Behavioral Intervention in Twins with Rett Syndrome. Journal of Autism and Developmental Disorders. 2019; 49: 5063-5072
12. Neul J.L, Lane J.B, Lee H, Geerts S, O Barrish J, Annese F et al. Developmental delay in Rett syndrome: data from the natural history study. Journal of Neurodevelopmental Disorders. 2014; 6: 20.
13. Banerjee A, Miller MT, Li K, Sur M, Kaufmann WE. Towards a better diagnosis and treatment of Rett syndrome: a model synaptic disorder. Brain: A Jornal of Neurology. 2019; 142: 239-248.
14. Vignoli A, Savini MN, Nowbut MS, Peron A, Turner K, La Briola F et al. Effectiveness and tolerability of antiepileptic drugs in 104 girls with Rett syndrome. Epilepsy & Behavior. 2017; 6: 27-33.
15. Van de Berg R, Breet LHM, Hiemstra M, Wagter L, Smeets E, Widdershoven J. Oculomotor Function in Individuals With Rett Syndrome. Elsevier: Pediatric Neurology. 2018; 88: 48-58.
16. Urbanowicz A, Downs J, Girdler S, Ciccone N, Leonarda H. An Exploration of the Use of Eye Gaze and Gestures in Females with Rett Syndrome. Journal of Speech, Language, and Hearing Research. 2016; 59: 1373-1383.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()