Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 17(supl 1)(1) - 2017
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Artigos de Revisao
Abordagem da cetoacidose diabética na infância e adolescência
Diabetes ketoacidosis approach in childhood and adolescence
Karina de Ferran1; Isla Aguiar Paiva2
1. Médica-Pediatra no Instituto de Puericultura e Pediatria Martagao Gesteira da Universidade Federal do Rio de Janeiro. Especialista em Endocrinologia Pediátrica pelo Instituto de Puericultura e Pediatria Martagao Gesteira da Universidade Federal do Rio de Janeiro. Mestre em Endocrinologia pela Universidade Federal do Rio de Janeiro
2. Médica-Pediatra no Instituto de Puericultura e Pediatria Martagao Gesteira da Universidade Federal do Rio de Janeiro. Especialista em Endocrinologia Pediátrica pelo Instituto de Puericultura e Pediatria Martagao Gesteira da Universidade Federal do Rio de Janeiro. Mestre em Endocrinologia pela Universidade Federal do Rio de Janeiro
Endereço para correspondência:
Recebido em: 13/9/2017 Aprovado em: 20/9/2017 Instituiçao: Universidade Federal do Rio de Janeiro
Resumo
INTRODUÇÃO: a cetoacidose diabética é a urgência endócrino-metabólica mais comum na infância e adolescência e a principal causa de hospitalização e óbito de pacientes diabéticos nessa faixa etária.
OBJETIVO: revisar a fisiopatologia da doença e propor uma abordagem diagnóstica e terapêutica para crianças e adolescentes, além de discutir medidas de prevenção.
FONTE DE DADOS: revisão não sistemática da literatura médica nacional e internacional de trabalhos originais, protocolos terapêuticos locais e consensos sobre o tema disponíveis até 2017, nas línguas portuguesa e inglesa, a partir de busca pelo PubMed.
SÍNTESE DE DADOS: a cetoacidose diabética pode ser a primeira manifestação clínica percebida na abertura de quadro de diabetes mellitus do tipo 1 ou ocorrer em paciente previamente diabético, por descontrole da doença. Os pilares do tratamento da cetoacidose diabética são a reposição de fluidos e insulina, objetivando a correção da acidose e dos distúrbios hidroeletrolíticos. A mortalidade relacionada à doença varia de 0,15 a 0,3%, sendo o edema cerebral a principal causa.
CONCLUSÃO: o diagnóstico precoce do diabetes mellitus do tipo 1 e o reconhecimento e manejo adequado da cetoacidose diabética são fundamentais para a redução da morbimortalidade do diabetes na infância.
Palavras-chave: Diabetes mellitus; Cetoacidose diabética; Edema encefálico.
Abstract
INTRODUCTION: diabetic ketoacidosis is the most common endocrine-metabolic urgency in childhood and adolescence and the main cause of hospitalization and death of diabetic patients in this age group.
OBJECTIVE: to review the pathophysiology of diabetic ketoacidosis and propose a diagnostic and therapeutic approach for children and adolescents, in addition to discuss prevention measures.
DATA SOURCE: non-systematic review of the national and international medical literature, original papers, Brazilian therapeutic protocols and consensus on the theme available until 2017, in the Portuguese and English languages, from PubMed search.
DATA SYNTHESIS: diabetic ketoacidosis may be the first clinical manifestation in newly diagnosed cases of type 1 diabetes mellitus or occurs in established type 1 diabetes, usually due to poor glycemic control or insulin omission. The pillars of the treatment of diabetic ketoacidosis are the replacement of fluids and insulin, aiming at the correction of acidosis and hydroelectrolytic disorders. Diabetic ketoacidosis related mortality ranges from 0.15 to 0.3%, with cerebral edema being the main cause.
CONCLUSION: the early diagnosis of type 1 diabetes mellitus and the adequate recognition and management of diabetic ketoacidosis are fundamental for reducing the morbidity and mortality of diabetes in childhood.
Keywords: Diabetes mellitus; Diabetic ketoacidosis; Brain edema.
INTRODUÇÃO
O diabetes mellitus do tipo 1 (DM1) é a doença endócrina mais comum da infância, acometendo mundialmente cerca de 65.000 crianças menores de 15 anos por ano.1 O quadro de cetoacidose diabética (CAD) é a urgência endócrino-metabólica mais comum nessa faixa etária e a principal causa de hospitalização e óbito de pacientes diabéticos na infância.
MÉTODO
Revisão não sistemática da literatura médica nacional e internacional de trabalhos originais, protocolos terapêuticos locais e consensos sobre o tema disponíveis até 2017, nas línguas portuguesa e inglesa, a partir de busca pelo PubMed. Os principais documentos utilizados para basear as recomendações apresentadas foram os consensos da American Diabetes Association e da International Society for Pediatric and Adolescent Diabetes.
RESULTADOS
A incidência de CAD na abertura do quadro de diabetes (como primeira manifestação da doença) varia muito geograficamente, com frequência de 11 a 67% em diferentes países europeus1,2 e até 80% em outras partes do mundo3 e é inversamente relacionada à incidência local de DM1.2 Acredita-se que quanto maior a incidência da doença, maior o conhecimento dos seus sinais e sintomas pela população e assim mais precoce a suspeição e o diagnóstico de DM1,3 antes da instalação da CAD. Além disso, a prevalência de CAD reduz significativamente o aumento da idade, como observado em estudo feito nos Estados Unidos,4 que mostrou 36% de prevalência de CAD em menores de 5 anos e 16% em maiores de 14 anos. Em crianças menores é difícil a percepção dos sintomas clássicos de poliúria, polidipsia e perda de peso, ocorrendo com maior frequência atraso no diagnóstico de DM1. A confusão do quadro de CAD com pneumonia ou asma, devido à taquipneia, ou com gastroenterite aguda e/ou abdome agudo, devido à presença de vômitos e dor abdominal,5 posterga o diagnóstico e leva ao agravamento dos sintomas, sendo frequente o diagnóstico de DM1 já num estágio avançado, com CAD grave.
A CAD ocorre também nos pacientes sabidamente diabéticos, com risco de 1 a 10% por paciente-ano.3 Esse risco é aumentado nos pacientes com omissão de doses de insulina, infecções, transgressão alimentar, controle metabólico ruim, história prévia de CAD, gastroenterite aguda com desidratação, doenças psiquiátricas, incluindo distúrbios alimentares, problemas sociais ou familiares, meninas adolescentes, acesso limitado aos serviços médicos e uso de bomba de insulina.6-8
A mortalidade das crianças com CAD varia de 0,15 a 0,3%, sendo o edema cerebral a principal causa, com incidência de 0,5 a 1,0% dos casos de CAD e mortalidade global de 60 a 90% de todas as mortes relacionadas à CAD na infância. A mortalidade é maior em locais onde os serviços de saúde têm menos estrutura.5 A fisiopatologia da doença é pouco conhecida. Em algumas coortes, foi associada à menor pressão parcial sanguínea de CO2 (pCO2), níveis mais elevados de ureia e ao uso de bicarbonato durante o tratamento da CAD.9,10
FISIOPATOGENIA
A CAD se deve ao estado de insulinopenia grave. Este pode decorrer da falência das células pancreáticas ou da falha na administração exógena da insulina, ou ainda da ineficácia de ação da insulina circulante devido ao antagonismo exercido por hormônios contrarreguladores, que promovem gliconeogênese e glicogenólise, além de limitar a utilização de glicose pelos tecidos periféricos, em situações de estresse metabólico, como na sepse, por exemplo.5
A hiperglicemia gerada promove diurese osmótica e subsequente desidratação e perda de eletrólitos pela urina, com hipoperfusão tissular e redução da taxa de filtração glomerular. Paralelamente, há elevação dos hormônios contrarreguladores, que, não contrapostos pela insulina, promovem proteólise com gliconeogênese, glicogenólise, redução da utilização periférica de glicose e lipólise. Os três primeiros contribuem para o aumento da glicemia, enquanto o último viabiliza a produção de ácidos graxos livres e de corpos cetônicos (cetogênese) - acetoacetato e β-hidroxibutirato. Esses cetoácidos consomem o sistema tampão de bases do organismo e causam acidose metabólica. A acidemia lática proporcionada pela desidratação e hipoperfusão tissular agrava a acidose metabólica.
Desidratação progressiva (por diurese osmótica e vômitos), acidose metabólica, distúrbios eletrolíticos e hiperosmolaridade estimulam ainda mais a liberação dos hormônios contrarreguladores do estresse, que atuam perpetuando o quadro e criando um ciclo vicioso, só quebrado quando o tratamento adequado é instaurado.5
DEFINIÇÃO DE CAD
O paciente pode apresentar hiperglicemia por diversos motivos e não preencher os critérios de CAD11 (Tabela 1). De forma oposta, o quadro de CAD pode estar instalado na ausência de hiperglicemia, situação que deve ser suspeitada em pacientes previamente diabéticos, que podem ter usado insulina pouco antes da chegada ao atendimento médico, numa tentativa de correção aguda da glicemia, mas insuficiente para a correção da acidose.

A gravidade da CAD é classificada pelo grau de acidose como leve, se pH menor que 7,3 ou bicarbonato menor que 15 mmol/l; moderada, se pH menor que 7,2, bicarbonato menor que 10 mmol/l; ou grave, se pH menor que 7,1, bicarbonato menor que 5 mmol/l.
ABORDAGEM AO PACIENTE COM CAD SUSPEITA
Uma boa anamnese e um exame físico completo devem ser realizados nos pacientes com suspeita de CAD, sendo estes sabidamente diabéticos ou não. Os exames laboratoriais são então colhidos para confirmação da suspeita diagnóstica e posterior acompanhamento do tratamento da CAD e dos possíveis fatores desencadeantes.
Anamnese e Exame Físico
No paciente sem diagnóstico prévio de DM1, buscar história de pólis (poliúria, polidipsia, polifagia) e emagrecimento recente, história pessoal ou familiar de autoimunidade, presença de acantose nigricans, obesidade e sinais e sintomas de intercorrência infecciosa. Já se o paciente é previamente diabético, investigar infecções intercorrentes, omissão de doses de insulina, falha no ajuste de doses durante intercorrência infecciosa, controle glicêmico anterior e eventos prévios de CAD.
Ao exame físico, sempre que possível, o paciente deve ser pesado. Além disso, deve ser avaliado quanto ao nível de consciência, à frequência e ao ritmo da respiração, ao grau de desidratação, à presença de sinais de infecção e deve ser realizada a palpação cuidadosa do abdome se houver queixa de dor abdominal. A avaliação periódica do nível de consciência pela escala de Glasgow12 é uma ferramenta útil durante todo o tratamento, possibilitando a detecção precoce de edema cerebral. A tabela 2 lista os principais sinais e sintomas de CAD.

Polidipsia e poliúria podem ser de difícil detecção em lactentes, considerando a falta de livre acesso à água (não verbaliza a sede) e o uso de fraldas.
A hiperventilação, provocada pela tentativa de correção da acidose metabólica pela alcalose respiratória, pode levar à impressão de que a taquipneia se deve a uma doença do aparelho respiratório, como pneumonia ou asma. A ausência de ruídos adventícios respiratórios deve alertar para o diagnóstico diferencial de CAD.
Vômitos e dor abdominal consequentes à acidose podem levar à confusão com gastroenterite aguda (atentar para a ausência de diarreia) e abdome agudo (Tabela 3). O hálito cetônico, descrito como de "maçã apodrecendo", pode estar presente.

Monitorização Clínica e Laboratorial no Tratamento de CAD
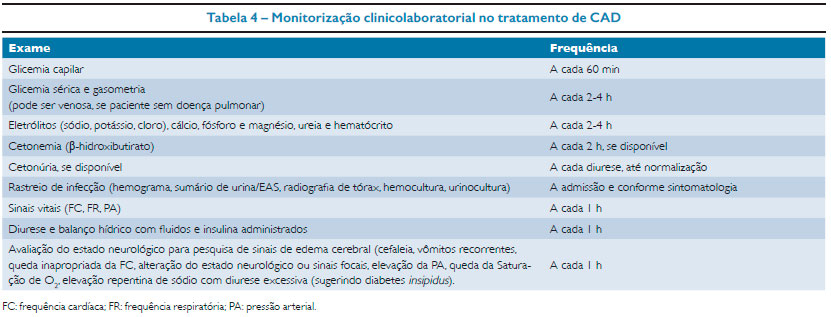
Uma vez confirmada a suspeita de CAD pela identificação dos critérios diagnósticos (Tabela 1), uma rotina de monitorização deve ser instituída durante o tratamento (Tabela 4).
O paciente deve ser tratado em unidade que tenha pessoal treinado no monitoramento e condução de crianças e adolescentes com CAD, com guias terapêuticos bem-estabelecidos e acesso a laboratório que forneça resultados dos exames pertinentes rapidamente.
Sempre que possível, um especialista no tratamento da CAD deve conduzir o tratamento do paciente, mesmo que remotamente. Isso se torna ainda mais importante se o paciente é atendido em um local com menos recursos e maior distância dos centros de referência.
As crianças que têm CAD mais grave (duração prolongada dos sintomas, choque e depressão do sensório) ou aquelas com risco aumentado de edema cerebral (idade < 5 anos, acidose grave, pCO2 baixo e ureia elevada) devem ser admitidas o mais rapidamente possível em unidade de terapia intensiva (UTI), preferencialmente pediátrica, ou em uma unidade que tenha recursos equivalentes e experiência no cuidado com diabéticos.11,13
Dois acessos venosos periféricos são necessários, pois idealmente a insulina deve ser infundida em via exclusiva, sendo a segunda via utilizada para hidratação venosa e coleta de exames. A via arterial é dispensável, exceto nos pacientes gravemente enfermos. Deve-se ainda evitar usar acesso venoso profundo pelo risco aumentado de trombose venosa, especialmente nas crianças menores. Se necessário seu uso, ele deve ser removido o mais precocemente possível.14 O cateterismo vesical não costuma ser necessário, salvo em pacientes inconscientes e com impossibilidade de urinar sob demanda (lactentes e pacientes gravemente enfermos). A monitorização eletrocardiográfica contínua é útil para avaliar sinais de hipo ou hipercalemia, a partir das alterações da onda T.15
Amostras de sangue periférico devem ser colhidas periodicamente para dosagem de glicemia, eletrólitos (sódio, potássio, cálcio, fósforo, magnésio), gasometria, ureia, creatinina, hemograma e corpos cetônicos (não disponíveis facilmente no país). A análise da urina deve ser feita para pesquisa de corpos cetônicos.
O paciente deve ser monitorizado detalhadamente em relação à sua resposta clínica e laboratorial. Deve-se registrar em uma folha de controle, de hora em hora, o estado clínico do paciente, as medicações venosas e orais, os líquidos administrados e os resultados dos exames laboratoriais realizados. Na tabela 4 estão descritos os intervalos de monitorização recomendados nos diferentes parâmetros analisados.16
Algumas considerações sobre os distúrbios hidroeletrolíticos e acidobásicos na CAD:
a) anion gap (AG) - tipicamente é aumentado na CAD devido à presença dos cetoácidos (entre 20 a 30 nmol/l). Valores de AG maiores que 35 nmol/l sugerem acidemia lática concomitante. O cálculo do AG é feito pela fórmula: AG = Na+ - (Cl- + HCO3). É considerado normal o AG = 12 ± 2 mmol/l;
b) osmolaridade plasmática (Osm) efetiva - é calculada pela fórmula Osm = 2 × Na + glicose/18. Normalmente ela está aumentada na CAD, entre 300 a 350 mOsm/l;17
c) sódio - na CAD ocorre hiponatremia dilucional: a cada 100 mg/dl de aumento da glicemia acima de 100 mg/dl, há uma queda de 1,6 mEq/l do sódio (Na+). O nível real de sódio plasmático é estimado pelo cálculo do Na+ corrigido pela fórmula: Na+ medido + 1,6 × (glicemia - 100)/100. Pode ainda ocorrer a pseudo-hiponatremia, em que a hiperlipidemia diminui a mensuração do sódio, mas as técnicas modernas de aferição do sódio minimizam este fenômeno. À medida que o paciente melhora e a glicemia cai, o sódio tende a subir, o que não indica piora da osmolaridade real. A queda do sódio ou seu aumento muito rápido (por diabetes insipidus) podem indicar edema cerebral.9
TRATAMENTO DA CAD
Os pilares do tratamento da CAD são a reposição de fluidos e de insulina, objetivando a correção da acidose e dos distúrbios hidroeletrolíticos. A restauração da perfusão tissular melhora a captação de glicose na periferia e a filtração glomerular; a insulinização reverte a proteólise, a lipólise e a cetogênese e estimula a captação e metabolização da glicose pelos tecidos, restaurando o metabolismo celular e normalizando a glicemia sérica.18 Todos esses efeitos contribuem para a correção da acidose.
Primeiramente, caso seja necessária a reanimação do paciente, devem ser seguidas as diretrizes do Pediatric Advanced Life Support (PALS), da American Heart Association (AHA), endossadas pela Sociedade Brasileira de Pediatria (SBP). No paciente inconsciente ou obnubilado, uma via aérea segura deve ser obtida e o estômago deve ser esvaziado por sonda nasogástrica (SNG) para evitar broncoaspiração. A intubação traqueal deve ser evitada, pois a subida rápida do pCO2 pode resultar em queda do pH do líquor e piorar o edema cerebral.19 Pacientes em choque hipovolêmico devem receber suplementação de oxigênio.
O uso de antibióticos é indicado para os pacientes com história de febre e deve ser iniciado após a coleta de culturas. O aumento dos leucócitos (leucocitose com desvio à esquerda) em resposta ao estresse é característico da CAD e não indica infecção.
Reposição de Líquidos
O deficit de fluidos extracelulares é de difícil estimativa e geralmente se encontra entre 5 a 10% do peso corporal.20,21
Geralmente estima-se que a CAD moderada tem deficit de líquidos de 5 a 7% e que a CAD grave tem deficit de 8 a 10% do peso corporal. O aumento da ureia e do hematócrito são indicadores do grau de depleção do líquido extracelular.22,23
Não existe consenso sobre qual esquema de reposição hidroeletrolítica é o mais seguro para evitar edema cerebral, portanto as recomendações a seguir se baseiam na decisão entre painel de especialistas da Lawson Wilkins Pediatric Endocrine Society (LWPES), da European Society for Paediatric Endocrinology (ESPE) e da (ISPAD), publicado em 2014,16 e ainda no protocolo utilizado pelo serviço de Diabetes Infantojuvenil no Instituto de Pediatria e Puericultura Martagão Gesteira da Universidade Federal do Rio de Janeiro (IPPMG/UFRJ) há mais de 20 anos.25,26
Os líquidos oferecidos desde o primeiro atendimento do paciente devem ser contabilizados no cálculo dos deficit e perdas.
Primeira fase: expansão volêmica
Para os pacientes com depleção de volume acentuada, mas sem choque, a expansão volêmica (ressuscitação) deve começar imediatamente com soro fisiológico (SF) a 0,9% intravenoso, visando restaurar a circulação periférica. O volume habitualmente administrado é de 10 a 20 ml/kg em uma a duas horas e pode ser repetido até melhora da perfusão tissular.
Nos raros casos com choque, deve ser realizada rápida expansão com SF 0,9% 20 ml/kg em bólus, visando restaurar o volume circulatório. Após cada etapa, o paciente deve ser reavaliado quanto a seu status circulatório e novas etapas de SF devem ser prescritas, conforme necessidade.
É recomendado que a expansão volêmica seja feita com soluções cristaloides. Não há dados que corroborem o uso de coloides em detrimento dos cristaloides na CAD.
Segunda fase: reposição de perdas hídricas e hidratação de manutenção
Nos pacientes com CAD leve, a fase de expansão pode não ser necessária, passando-se diretamente para a fase de reposição das perdas e manutenção.
Durante o tratamento, à medida que a glicemia cai, há queda da osmolaridade plasmática e consequentemente da volemia. É essencial que o paciente esteja recebendo líquidos e sódio suficientes para manter a adequada perfusão tissular.
Além de fornecer a necessidade hídrica diária (NHD), as perdas devem ser estimadas e repostas em 48 horas.11,23,27 Normalmente, entretanto, os pacientes já estão melhores da CAD e em condições de ingerir por via oral já nas primeiras 24 horas de tratamento,27 sendo o tempo médio de saída da CAD de 11,6 ± 6,2 horas, segundo estudo que analisou 635 episódios de CAD. Assim, o restante da hidratação planejada para reposição de perdas em 48 horas, pode ser oferecida pela via oral, juntamente com a insulina subcutânea (SC).27
O cálculo da necessidade hídrica diária pode ser feito de várias formas, mas utilizamos o método de Holiday Segar e optamos por subestimar as perdas e repô-las em 24 horas.25,26
Cálculo da necessidade hídrica diária (NHD) pelo método de Holiday Segar:
a) até 10 kg de peso - 100 ml/kg/dia;
b) 11 a 20 kg de peso - 1.000 ml + (50 ml/kg do peso que exceder 10 kg) no dia;
c) 21 a 45 kg de peso - 1.500 ml + (20 ml/kg do peso que exceder 20 kg) no dia;
d) acima de 45 kg de peso - 2.000 ml/dia.
Cálculo das perdas de acordo com o grau de desidratação, tendendo a subestimá-las para repor em 24 horas: 25,26
a) leve (3 a 5% do peso)- 30 a 50 ml/kg;
b) moderada (5 a 7% do peso)- 50 a 70 ml/kg;
c) grave (não ultrapassar 10% do peso)- 80 a 100 ml/kg.
O volume total calculado para 24 horas (NHD + perdas) é dividido em três partes, sendo ⅓ infundido nas primeiras seis horas, ⅓ infundido nas seis horas seguintes e o último terço infundido nas últimas 12 horas do dia. Deve-se ter o cuidado para não ultrapassar o volume máximo de 4 litros/m2/dia.
Inicialmente, deve-se usar soluções isotônicas (SF 0,9% ou ringer lactato) por quatro a seis horas.23,28,29 A glicose deverá ser adicionada à solução quando a glicemia atingir valores entre 250 e 300 mg/dl, na proporção de 1:1 (volumes iguais de SF 0,9% e de soro glicosado a 5%), também chamada de solução glicofisiológica.
Quando a glicemia for menor que 120 mg/dl, deve-se trocar a hidratação para SG a 5% ou até com maiores concentrações de glicose, aumentando a taxa de infusão de glicose (TIG), visando manter a glicemia maior que 120 mg/dl. O sódio deverá ser adicionado a esta solução, visando manter a osmolaridade mais elevada, em torno de 150 mOsm/l (equivalente a do SF 0,45%). As perdas urinárias não devem ser repostas rotineiramente.
O uso de grandes quantidades de fluidos ricos em cloreto pode estar associado ao desenvolvimento de hipercloremia (relação Cl/Na > 0,7930,31 e acidose metabólica hiperclorêmica concomitante).32,33 Isso pode mascarar a resolução da CAD,30 mantendo um deficit de bases ou um bicarbonato persistentemente baixo. A dosagem sérica de corpos cetônicos pode ser útil na diferenciação dos dois quadros. A acidose hiperclorêmica se resolve espontaneamente. O excesso de cloretos pode ser evitado usando soluções isotônicas, como ringer lactato.
Insulinização
Apesar da hidratação venosa promover queda da glicemia, a insulinização é essencial para restaurar o metabolismo celular normal, suprimir lipólise, proteólise e cetogênese e, assim, normalizar a glicemia e contribuir na reversão da acidose.34
A insulina deve ser iniciada após término da expansão volêmica, ao final da primeira hora de tratamento.52
Não se utiliza mais a dose de ataque de insulina antes do início da infusão contínua, pois pode levar a uma rápida queda da glicemia e da osmolaridade sérica, o que aumenta o risco de edema cerebral,35,36 além de poder agravar a hipocalemia.
A insulinização deve ser feita com infusão contínua venosa de insulina regular, na dose de 0,1 unidade/kg/hora. Deve-se saturar o equipo com 50 ml da solução, desprezando esse volume e repetindo o procedimento a cada troca de solução, o que deve ocorrer a cada seis horas.
Visando facilitar cálculos, sugerimos a solução preparada com 30 unidades de insulina regular mais 300 ml de SF 0,9% (0,1 unidade de insulina por ml de solução). Com essa solução, a dose de 0,1 unidade/kg/h de insulina corresponde a 1 ml/kg/h de solução, ou seja, basta colocar o peso da criança na velocidade da bomba infusora (por exemplo, um paciente de 20 kg receberá essa solução em infusão a 20 ml/h).
A infusão contínua deve ser mantida até resolução da CAD (pH > 7,3 e bicarbonato > 15 nmol/l), o que sempre vai ocorrer após a normalização da glicemia.37
Alguns centros utilizam rotineiramente doses menores de insulina para prevenir a hipoglicemia (0,05 unidade/kg/h),38 baseados em estudos observacionais retrospectivos que relataram eficácia semelhante entre as duas doses. Não existem estudos randomizados controlados comparativos e não existe evidência de que a dose de 0,1 unidade/kg/h seja danosa.
A insulina tem efeito semelhante ao da aldosterona, levando a aumento da excreção renal de potássio. Altas doses de insulina administradas prolongadamente podem contribuir para redução dos níveis séricos de potássio, apesar da sua reposição venosa. Assim, o tempo e a dose de insulina infundidos devem ser minimizados para evitar hipocalemia severa.39
Durante a infusão contínua de insulina, espera-se que a glicose caia cerca de 40 a 90 mg/dl por hora. Caso haja uma queda da glicemia maior que 90 mg/dl/h,40,41 a glicose pode ser acrescentada na hidratação venosa, mesmo antes da glicemia estar menor que 300 mg/dl.
Caso os parâmetros laboratoriais não melhorem, o paciente deve ser reavaliado, as doses e soluções de insulina e soro devem ser recalculadas e checadas, avaliando se o preparo está adequado e potenciais infecções não diagnosticadas/tratadas devem ser consideradas.
Reposição de Eletrólitos
O paciente com CAD tem redução do conteúdo total de potássio (K+) corporal, na ordem de 3 a 6 mmol/kg.20,21 O aumento da osmolaridade plasmática, a glicogenólise, a proteólise e a acidose metabólica causam saída do potássio intracelular para o extracelular. Os vômitos e a diurese osmótica promovem a perda do potássio corporal. A desidratação resulta em hiperaldosteronismo secundário com aumento da excreção renal de potássio. Assim, diversos mecanismos atuam para reduzir o conteúdo total de potássio corporal, apesar de poder haver hipercalemia no início do tratamento, especialmente nos casos de disfunção renal, decorrente da hipoperfusão tissular, que reduz excreção de potássio e agrava a acidose.42 A insulinização e a correção da acidose promovem retorno do potássio para o meio intracelular, com rápida queda dos seus níveis séricos,43 predispondo o paciente a arritmias cardíacas. A reposição do potássio é necessária independentemente dos níveis séricos, salvo na presença de insuficiência renal.44
O eletrocardiograma (ECG) pode auxiliar na determinação de hiper ou hipocalemia.15
A reposição de potássio deve ser iniciada assim que a diurese for reestabelecida:26
a) se K+ maior que 6 mEq/l - aguardar queda para iniciar reposição;
b) se K+ maior ou igual a 4,5 mEq/l - repor 20 mEq de potássio por litro de solução;
c) se K+ menor que 4,5 mEq/l - repor 40 mEq de potássio por litro de solução.
O fosfato intracelular também está depletado na CAD e é perdido pela diurese osmótica.20,21 Os níveis séricos caem após início do tratamento, com entrada do fosfato para dentro das células.45 Hipofosfatemia clinicamente significativa pode ocorrer se houver jejum prolongado (> 24 h).20,21 Estudos com amostras reduzidas não mostraram benefício na reposição de fósforo.46
Hipofosfatemia grave combinada com depleção de fosfato pode ter consequências significativas, porém é bastante rara. As manifestações clínicas dependem da gravidade e cronicidade da depleção de fosfato, não havendo sintomas até serem atingidos níveis plasmáticos menores que 1 mg/dl.
No tratamento da CAD, hipofosfatemia grave pode ocorrer, mas o aparecimento de sintomas é incomum porque geralmente o quadro é agudo e não há passado de depleção crônica de fosfato. Só recomendamos a reposição de fosfato para os pacientes com hipofosfatemia grave que apresentem patologia pulmonar concomitante que possa cursar com hipoxemia e nos casos de jejum prolongado de mais de 48 horas após o tratamento. A reposição de fosfato pode levar à hipocalcemia.
O uso de bicarbonato não está mais indicado na CAD, independentemente dos níveis de pH. Estudos controlados mostraram não haver benefício.47 A terapia com bicarbonato pode causar acidose paradoxal no sistema nervoso central48 e hipocalemia grave,48 além de estar associada, em alguns estudos, ao aumento do risco de edema cerebral.9 Atualmente, seu uso só está indicado na parada cardíaca.
COMPLICAÇÕES DURANTE O TRATAMENTO DA CAD
As complicações observadas durante o tratamento da CAD são bastante graves e devem ser monitorizadas e tratadas prontamente.
Hipoglicemia
Hipoglicemia é definida como glicemia abaixo de 60 mg/dl em crianças maiores de 1 ano de idade. Em caso de hipoglicemia durante a correção de CAD, fazer flush de 200 mg/kg de glicose usando, por exemplo, 2 ml/kg de soro glicosado a 10%. Em seguida, aumentar concentração do soro glicosado (ou a TIG) da hidratação venosa em uso.
Edema Cerebral
O edema cerebral é a complicação mais temida do tratamento da CAD. Tipicamente ocorre 4 a 12 horas após início do tratamento, mas pode ocorrer mesmo antes dele ter sido instituído ou mais tardiamente, como após 24 horas de iniciado.16
O edema cerebral franco é uma complicação rara, ocorrendo em 0,5 a 0,9% dos episódios de CAD, mas de alta mortalidade (21-24%), sendo a principal causa de óbito em crianças com CAD, além de responsável por grande taxa de sobreviventes com sequela neurológica permanente (15-26%).9,10
Estudos observaram que aproximadamente 15% dos pacientes tratados para CAD tinham alteração do nível de consciência (Glasgow < 14) com evidência de edema cerebral na neuroimagem.49 Outros, de forma semelhante, evidenciaram que o edema cerebral na neuroimagem é fenômeno relativamente frequente,49 com graus variados, sendo o edema cerebral clinicamente evidente uma manifestação grave de um fenômeno comum.50
A etiologia do edema cerebral permanece não esclarecida completamente e as hipóteses atribuindo sua fisiopatogenia à infusão rápida de fluidos com variação brusca da osmolaridade ainda são controversas. Este dado é corroborado por investigações mais recentes que atribuíram a desidratação e a hipoperfusão cerebral geradas pela CAD à injúria cerebral,9,51 sugerindo que fatores intrínsecos à CAD podem levar à lesão cerebral e esta pode ser agravada durante o tratamento.52 De fato, o grau de edema que se desenvolve durante a CAD se correlaciona ao grau de desidratação e hiperventilação à admissão, mas não com a osmolaridade inicial ou mudanças de osmolaridade ao longo do tratamento.
Os fatores de risco que já foram associados ao edema cerebral sintomático são: idade menor de 5 anos ;53 abertura de quadro de DM1;53 maior duração dos sintomas e, à admissão, pCO2 proporcionalmente mais baixa do que o esperado para a acidose;9 ureia aumentada;9 acidose muito grave35 e durante tratamento, o uso de bicarbonato;9 grandes volumes infundidos nas primeiras quatro horas;35 queda rápida da osmolaridade sérica;36 administração de insulina na primeira hora de reposição de fluidos;35 e uma elevação atenuada do sódio ou uma queda do sódio corrigido durante o tratamento.9
Uma abordagem baseada na avaliação periódica do exame neurológico à beira do leito permite a identificação de sinais que podem ser usados como critérios diagnósticos, critérios maiores e critérios menores para edema cerebral.54 A presença de um critério diagnóstico ou dois critérios maiores ou um critério maior e dois critérios menores (Tabela 5), após o início do tratamento, tem sensibilidade de 92% para o diagnóstico de edema cerebral, com apenas 4% de falso-positivo, e é sugerida pelo guideline mais recente.16

Na suspeita clínica de edema cerebral, o tratamento deve ser iniciado prontamente, antes mesmo da realização de exames de imagem. As medidas preconizadas são: reduzir a velocidade de infusão de fluidos a ⅓ da vigente; elevar a cabeceira a 30° e administrar manitol na dose de 0,5 a 1,0 g/kg, intravenoso, em 10 a 15 minutos, repetindo a dose se não houver resposta inicial em 30 minutos a 2 horas.55 A salina hipertônica (3%), na dose de 2,5 a 5 ml/kg, em infusão por 10 a 15 minutos, pode ser usada como alternativa ao manitol, especialmente se não houver resposta inicial ao manitol, porém estudos sugerem que ela pode não ser tão eficaz quanto o manitol e estar associada a maior mortalidade quando usada como primeira escolha.56 A intubação traqueal pode ser necessária para proteção de vias aéreas, mas deve ser evitada a hiperventilação, pois piora o desfecho neurológico. O exame de imagem do SNC deve ser realizado após iniciado o tratamento do edema cerebral, visando descartar diagnósticos diferenciais que requeiram outras abordagens, como neurocirurgia (hemorragia intracraniana, por exemplo) ou anticoagulação (trombose cerebrovascular, por exemplo).
FASE DE TRANSIÇÃO
Uma vez obtida a correção da cetoacidose, confirmada pelos níveis de pH maiores que 7,3 e bicarbonato maior que 15 mEq/l, associados à glicemia menor que 250 mg/dl, deve ser iniciada a insulinização subcutânea, para subsequente interrupção da infusão venosa de insulina.26
O ideal é que essa transição da insulinização venosa para subcutânea seja feita com o paciente acordado e disposto a receber uma refeição. Pode-se usar a insulina regular 30 minutos antes ou a insulina ultrarrápida (asparte, lispro ou glulisina) 15 minutos antes da suspensão da infusão venosa de insulina. A dose é de 0,1 UI/kg. 26 Em seguida, oferecer alimentos conforme horário. Neste momento também deve ser interrompida a infusão de hidratação venosa que contenha soro glicosado e a hidratação pode ser oferecida por via oral e/ou venosa (com SF 0,9% e potássio), mantendo-se os volumes previamente calculados. O tratamento com insulina regular ou ultrarrápida por via subcutânea é mantido e as doses escolhidas de acordo com as glicemias capilares (hemoglucoteste) antes das refeições. Na tabela 6 segue uma sugestão de posologia.
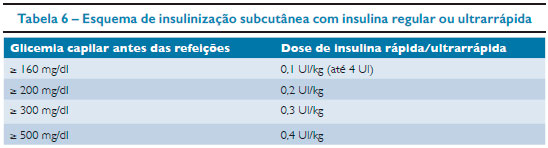
Recomenda-se que sejam usadas doses menores de insulina regular/ultrarrápida antes da ceia (somente se glicemia acima de 200 mg/dl). Idealmente, concomitantemente à suspensão da infusão contínua de insulina, deve ser iniciada a insulinização subcutânea basal, com insulina lenta ou ultralenta (NPH ou glargina), tentando-se ajustar o horário de aplicação para antes do café da manhã ou da ceia. A dose inicial da insulina NPH é 0,3 a 0,5 UI/kg/dia, fracionado em ⅔ pela manhã e ⅓ na ceia, e a de insulina glargina é 0,2 a 0,4 UI/kg/dia, em tomada única.26
Em caso de hipoglicemia nessa fase, no paciente assintomático, recomenda-se oferecer pequeno lanche (200 ml de leite com pão) ou antecipar a refeição prevista para o horário. Caso o paciente esteja sintomático, mas lúcido, ou se for o horário da ceia ou de madrugada, oferecer uma colher de sopa ou três sachês de açúcar (15 g de carboidrato simples), além de pequeno lanche ou da refeição do horário. Já se o paciente estiver sonolento ou inconsciente, administrar flush de glicose (como descrito acima) e oferecer lanche, caso o paciente tolere a via oral, ou aumentar/iniciar hidratação com soro glicosado, caso a via oral não seja possível. Nova glicemia capilar deve ser verificada após 30 a 60 minutos, repetindo-se os procedimentos acima caso não haja normalização da glicemia.
PREVENÇÃO
A abordagem à CAD só é completa quando é identificado o seu fator precipitante, possibilitando a orientação do paciente e de sua família quanto às medidas de prevenção de recorrência.
No paciente em abertura de quadro de DM1, o reconhecimento dos sinais e sintomas da doença, especialmente em lactentes e crianças pequenas, pode evitar a evolução para CAD. O paciente recém-diagnosticado com DM1 e seus cuidadores devem receber educação em diabetes antes da alta hospitalar, com treinamento para aferição de glicemia, aplicação de insulina, manejo de doses, orientação nutricional, além do reconhecimento dos sinais de hipoglicemia e de CAD.57
Já em pacientes previamente diabéticos, a principal causa de CAD é a omissão de dose de insulina, seja inadvertida ou deliberadamente. A supervisão por um adulto do uso de insulina, especialmente em adolescentes e em casos de estresse físico, infeccioso ou emocional, é medida importante na prevenção de CAD, capaz de reduzir em até dez vezes sua recorrência.58 Naqueles usuários de bomba de insulina, é comum CAD associada a falha no equipamento, que deve ser rotineiramente checado. O uso de fitas de monitorização de cetonemia pode auxiliar na detecção precoce da CAD.59
COMENTÁRIOS
O diagnóstico precoce do DM1 e o reconhecimento e manejo adequado da CAD são fundamentais para a redução da morbimortalidade do diabetes na infância.
AGRADECIMENTO
Agradecemos ao Dr. Jorge Luiz Luescher, chefe do Ambulatório de Diabetes Infantojuvenil do IPPMG/UFRJ, pelo apoio à publicação e pelos anos de ensinamentos e dedicação aos seus alunos e pacientes.
REFERÊNCIAS
1 DIAMOND Project Group. Incidence and trends of childhood type 1 diabetes worldwide 1990-1999. Diabet Med. 2006;23(8):857-66.
2 Lévy-Marchal C, Patterson CC, Green A; EURODIAB ACE Study Group. Geographical variation of presentation at diagnosis of type I diabetes in children: the EURODIAB study. European and Diabetes. Diabetologia. 2001;44(suppl 3):b75-80.
3 Usher-Smith JA, Thompson M, Ercole A, Walter FM. Variation between countries in the frequency of diabetic ketoacidosis at first presentation of type 1 diabetes in children: a systematic review. Diabetologia. 2012;55(11):2878-94.
4 Rewers A, Klingensmith G, Davis C, Petitti D, Pihoker C, Rodriguez B et al. The presence of diabetic ketoacidosis at diagnosis of diabetes mellitus in youth: the Search for Diabetes in Youth Study. Pediatrics. 2008;121(5):e1258-66.
5 Wolfsdorf J, Glaser N, Sperling MA; American Diabetes Association. Diabetic ketoacidosis in infants, children, and adolescents: a consensus statement from the American Diabetes Association. Diabetes Care. 2006;29(5):1150-9.
6 Rosilio M, Cotton JB, Wieliczko MC, Gendrault B, Carel JC, Couvaras O et al. Factors associated with glycemic control. A cross-sectional nationwide study in 2,579 French children with type 1 diabetes. The French Pediatric Diabetes Group. Diabetes Care. 1998;21:(7)1146-53.
7 Morris AD, Boyle DI, McMahon AD, Greene SA, MacDonald TM, Newton RW. Adherence to insulin treatment, glycaemic control, and ketoacidosis in insulin-dependent diabetes mellitus. The DARTS/MEMO Collaboration. Diabetes Audit and Research in Tayside Scotland. Medicines Monitoring Unit. Lancet. 1997;350(9090):1505-10.
8 Cengiz E, Xing D, Wong JC, Wolfsdorf JI, Haymond MW, Rewers A et al. Severe hypoglycemia and diabetic ketoacidosis among youth with type 1 diabetes in the T1D Exchange clinic registry. Pediatr Diabetes. 2013;14:(6)447-54.
9 Glaser N, Barnett P, McCaslin I, Nelson D, Trainor J, Louie J et al: Risk factors for cerebral edema in children with diabetic ketoacidosis: the Pediatric Emergency Medicine Collaborative Research Committee of the American Academy of Pediatrics. N Engl J Med. 2001;344(4):264-9.
10 Lawrence SE, Cummings EA, Gaboury I, Daneman D. Population-based study of incidence and risk factors for cerebral edema in pediatric diabetic ketoacidosis. J Pediatr. 2005;146(5):688-92.
11 Dunger DB, Sperling MA, Acerini CL, Bohn DJ, Daneman D, Danne TP et al. ESPE/LWPES consensus statement on diabetic ketoacidosis in children and adolescents. Arch Dis Child. 2004;89:(2)188-94.
12 Reilly PL, Simpson DA, Sprod R, Thomas L. Assessing the conscious level in infants and young children: a paediatric version of the Glasgow Coma Scale. Childs Nerv Syst. 1988;4(1):30-3.
13 Monroe KW, King W, Atchison JA. Use of PRISM scores in triage of pediatric patients with diabetic ketoacidosis. Am J Manag Care. 1997;3:(2)253-8.
14 Gutierrez JA, Bagatell R, Samson MP, Theodorou AA, Berg RA. Femoral central venous catheter associated deep venous thrombosis in children with diabetic ketoacidosis. Crit Care Med. 2003;31(1):80-3.
15. Malone JI, Brodsky SJ. The value of electrocardiogram monitoring in diabetic ketoacidosis. Diabetes Care. 1980;3(4):543-7.
16 Wolfsdorf JI, Allgrove J, Craig ME, Edge J, Glaser N, Jain V et al. A consensus statement from the international society for pediatric and adolescent diabetes: diabetic ketoacidosis and hyperglycemic hyperosmolar state. Pediatric Diabetes. 2014;15(suppl 20):154-79.
17 Halperin ML, Goldstein MB. Fluid, electrolyte, and acid-base physiology. 3th ed. Philadelphia: Saunders; 1999.
18 Rosenbloom AL. The management of diabetic ketoacidosis in children. Diabetes Ther. 2010;1(2):103-20.
19 Tasker RC, Lutman D, Peters MJ. Hyperventilation in severe diabetic ketoacidosis. Pediatr Crit Care Med. 2005;6(4):405-11.
20 Atchley D, Loeb R, Richards Jr D, Benedict E, Driscoll M. On diabetic ketoacidosis: a detailed study of electrolyte balances following the withdrawal and reestablishment of insulin therapy. J Clin Invest. 1933;12(2):297-326.
21 Nabarro J, Spencer AG, Stowers JM. Metabolic studies in severe diabetic ketosis. Q J Med. 1952;21(82):225-48.
22 Linares MY, Schunk JE, Lindsay R. Laboratory presentation in diabetic ketoacidosis and duration of therapy. Pediatr Emerg Care. 1996;12(5):347-51.
23 Harris GD, Fiordalisi I. Physiologic management of diabetic ketoacidemia. A 5-year prospective pediatric experience in 231 episodes. Arch Pediatr Adolesc Med. 1994;148(10):1046-52.
24 Dunger DB, Sperling MA, Acerini CL, Bohn DJ, Daneman D, Danne TP et al. European Society for Paediatric Endocrinology/Lawson Wilkins Pediatric Endocrine Society consensus statement on diabetic ketoacidosis in children and adolescents. Pediatrics. 2004:113(2):e133-40.
25 Bevilacqua CC, Moraes SRS, Fernandes MF, Costa AM, Rodrigues MCF. Emergências pediátricas do Instituto de Puericultura e Pediatria Martagão Gesteira - Universidade Federal do Rio de Janeiro - IPPMG/UFRJ. São Paulo: Editora Atheneu; 2000.
26 Azevedo CES. Bases da pediatria. Rio de Janeiro: Rubio; 2013.
27 Fiordalisi I, Novotny WE, Holbert D, Finberg L, Harris GD; Critical Care Management Group. An 18-yr prospective study of pediatric diabetic ketoacidosis: an approach to minimizing the risk of brain herniation during treatment. Pediatr Diabetes. 2007;8(3):142-9.
28 Mel JM, Werther GA. Incidence and outcome of diabetic cerebral oedema in childhood: are there predictors? J Paediatr Child Health. 1995;31(1):17-20.
29 Toledo JD, Modesto V, Peinador M, Alvarez P, López-Prats JL, Sanchis R et al. Sodium concentration in rehydration fluids for children with ketoacidotic diabetes: effect on serum sodium concentration. J Pediatr. 2009;154(6):895-900.
30 Taylor D, Durward A, Tibby SM, Thorburn K, Holton F, Johnstone IC et al. The influence of hyperchloraemia on acid base interpretation in diabetic ketoacidosis. Intensive Care Med. 2006;32(2):295-301.
31 Durward A, Skellett S, Mayer A, Taylor D, Tibby SM, Murdoch IA. The value of the chloride: sodium ratio in differentiating the aetiology of metabolic acidosis. Intensive Care Med. 2001;27(5):828-35.
32 Oh MS, Carroll HJ, Uribarri J. Mechanism of normochloremic and hyperchloremic acidosis in diabetic ketoacidosis. Nephron. 1990;54(1):1-6.
33 Basnet S, Venepalli PK, Andoh J, Verhulst S, Koirala J. Effect of normal saline and half normal saline on serum electrolytes during recovery phase of diabetic ketoacidosis. J Intensive Care Med. 2014;29(1):38-42.
34 Luzi L, Barrett EJ, Groop LC, Ferrannini E, DeFronzo RA. Metabolic effects of low-dose insulin therapy on glucose metabolism in diabetic ketoacidosis. Diabetes. 1988;37(11):1470-7.
35 Edge JA, Jakes RW, Roy Y, Hawkins M, Winter D, Ford-Adams ME et al. The UK case-control study of cerebral oedema complicating diabetic ketoacidosis in children. Diabetologia. 2006;49(9): 2002-9.
36 Hoorn EJ, Carlotti AP, Costa LA, MacMahon B, Bohn G, Zietse R et al. Preventing a drop in effective plasma osmolality to minimize the likelihood of cerebral edema during treatment of children with diabetic ketoacidosis. J Pediatr. 2007;150(5):467-73.
37 Soler NG, FitzGerald MG, Wright AD, Malins JM. Comparative study of different insulin regimens in management of diabetic ketoacidosis. Lancet. 1975;2(7947):1221-4.
38 Puttha R, Cooke D, Subbarayan A, Odeka E, Ariyawansa I, Bone M et al. Low dose (0.05 units/kg/h) is comparable with standard dose (0.1 units/kg/h) intravenous insulin infusion for the initial treatment of diabetic ketoacidosis in children with type 1 diabetes-an observational study. Pediatr Diabetes. 2010;11(1):12-7.
39 Carlotti AP, St George-Hyslop C, Bohn D, Halperin ML. Hypokalemia during treatment of diabetic ketoacidosis: clinical evidence for an aldosterone-like action of insulin. J Pediatr. 2013;163(1):207-12.e201.
40 Martin MM, Martin AA. Continuous low-dose infusion of insulin in the treatment of diabetic ketoacidosis in children. J Pediatr. 1976;89(4):560-4.
41 Kappy MS, Lightner ES. Low-dose intravenous insulin in the treatment of diabetic ketoacidosis. Am J Dis Child. 1979;133(5):523-5.
42 Adrogue HJ, Lederer ED, Suki WN, Eknoyan G. Determinants of plasma potassium levels in diabetic ketoacidosis. Medicine (Baltimore). 1986;65(3):163-72.
43 DeFronzo RA, Felig P, Ferrannini E, Wahren J. Effect of graded doses of insulin on esplanchnic and peripheral potassium metabolism in man. Am J Physiol. 1980;238(5):E421-7.
44 Tattersall RB. A paper which changed clinical practice (slowly). Jacob Holler on potassium deficiency in diabetic acidosis (1946). Diabet Med. 1999;16(21):978-84.
45 Riley MS, Schade DS, Eaton RP. Effects of insulin infusion on plasma phosphate in diabetic patients. Metabolism. 1979;28(3):191-4.
46 Clerbaux T, Reynaert M, Willems E, Frans A. Effect of phosphate on oxygen-hemoglobin affinity, diphosphoglycerate and blood gases during recovery from diabetic ketoacidosis. Intensive Care Med. 1989;15(8):495-8.
47 Green SM, Rothrock SG, Ho JD, Gallant RD, Borger R, Thomas TL et al. Failure of adjunctive bicarbonate to improve outcome in severe pediatric diabetic ketoacidosis. Ann Emerg Med. 1998;31(1):41-8.
48 Assal JP, Aoki TT, Manzano FM, Kozak GP. Metabolic effects of sodium bicarbonate in management of diabetic ketoacidosis. Diabetes. 1974;23(5):405-11.
49 Glaser NS, Wootton-Gorges SL, Buonocore MH, Marcin JP, Rewers A, Strain J et al. Frequency of sub-clinical cerebral edema in children with diabetic ketoacidosis. Pediatr Diabetes. 2006;7(2):75-80.
50 Sperling MA. Cerebral edema in diabetic ketoacidosis: an underestimated complication? Pediatr Diabetes. 2006;7(2):73-4.
51 Glaser NS, Wootton-Gorges SL, Marcin JP, Buonocore MH, Dicarlo J, Neely EK et al. Mechanism of cerebral edema in children with diabetic ketoacidosis. J Pediatr. 2004;145(2):164-71.
52 Glaser N. Cerebral injury and cerebral edema in children with diabetic ketoacidosis: could cerebral ischemia and reperfusion injury be involved? Pediatr Diabetes. 2009;10(8):534-41.
53 Rosenbloom AL. Intracerebral crises during treatment of diabetic ketoacidosis. Diabetes Care. 1990;13(1):22-33.
54 Muir AB, Quisling RG, Yang MC, Rosenbloom AL. Cerebral edema in childhood diabetic ketoacidosis: natural history, radiographic findings, and early identification. Diabetes Care. 2004:27(7):1541-6.
55 Roberts MD, Slover RH, Chase HP. Diabetic ketoacidosis with intracerebral complications. Pediatr Diabetes. 2001;2(3):109-14.
56 Decourcey DD, Steil GM, Wypij D, Agus MS. Increasing use of hypertonic saline over mannitol in the treatment of symptomatic cerebral edema in pediatric diabetic ketoacidosis: an 11-year retrospective analysis of mortality. Pediatr Crit Care Med. 2013;14(7):694-700.
57 Illinois Emergency Medical Services for Children. Pediatric Hyperglycemia and Diabetic Ketoacidosis (DKA). 5th ed. 2017. Available from: http://ssom.luc.edu/media/stritchschoolofmedicine/emergencymedicine/emsforchildren/documents/education/allhealthcareprofessionals/pedDKA_pdf.pdf
58 Golden MP, Herrold AJ, Orr DP. An approach to prevention of recurrent diabetic ketoacidosis in the pediatric population. J Pediatr. 1985;107(2):195-200.
59 Laffel LM, Wentzell K, Loughlin C, Tovar A, Moltz K, Brink S. Sick day management using blood 3-hydroxybutyrate (3-OHB) compared with urine ketone monitoring reduces hospital visits in young people with T1DM: a randomized clinical trial. Diabet Med. 2006;23(3):278-84.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()