Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 23(1) - Março 2023
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Miocardiopatia em irmãos como manifestação inicial de Síndrome de Alstrom: Relato de dois casos
Myocardiopathy in siblings as initial manifestation of Alstrom Syndrome: Report of two cases
Isadora de Oliveira Cavalcante1; Juliana Pastana Ramos de-Freitas1; Melina Rodero Marques1; Cristina Machado Camargo Afiune2; Paulo Emídio Lobão Cunha1; Jeanne Alves de Souza Mazza1; Lisiane Seguti Ferreira1
DOI:10.31365/issn.2595-1769.v23i1p09-11
1. Universidade de Brasília, Neurologia Infantil - Brasília - DF - Brasil
2. Instituto de Cardiologia do Distrito Federal, Cardiologia Pediátrica - Brasília - DF - Brasil
Endereço para correspondência:
Isadora de Oliveira Cavalcante
isadora.oc.med@gmail.com
Instituição: Universidade de Brasília, Neurologia Infantil - Brasília - DF - Brasil
Recebido em: 02/02/2022
Aprovado em: 08/04/2023
Resumo
INTRODUÇÃO: A síndrome de Alstrom é uma ciliopatia monogênica recessiva rara, de apresentação diversa, com dificuldade de diagnóstico precoce sobretudo em crianças menores de cinco anos.
OBJETIVOS: Este artigo objetiva relatar dois casos de síndrome de Alstrom em irmãos, tendo como manifestação inicial a cardiomiopatia dilatada. Relato de caso: São relatados dois casos da síndrome em um casal de irmãos, cuja sintomatologia inicial foi a cardiomiopatia dilatada, porém de evoluções distintas entre si, com recuperação total da cardiomiopatia em um deles. Diagnóstico realizado inicialmente na irmã mais velha, após procurar serviço de Neurologia Infantil devido a movimentos oculares horizontais, apresentando história pregressa de cardiomiopatia, evidenciando-se ainda obesidade e hipertrigliceridemia. Seu irmão apresentou também quadro de miocardiopatia dilatada, mas segue sem outros sintomas da síndrome. O diagnóstico de ambos foi confirmado após análise molecular.
DISCUSSÃO: Chama a atenção, neste relato, a variabilidade intrafamiliar da doença, tanto relacionada à sintomatologia apresentada, quanto à evolução, dificultando ainda mais o diagnóstico precoce da síndrome. Essa variabilidade sugere que fatores epigenéticos podem desempenhar um papel na apresentação da doença, principalmente por modificação alélica.
Palavras-chave: Cardiomiopatia dilatada. Síndrome de Alstrom. Irmãos.
Abstract
INTRODUCTION: Alstrom syndrome is a rare monogenic recessive ciliopathy, with different presentation, presenting difficulty in early diagnosis, especially in children under five years old.
OBJECTIVES: This paper aims to report two cases of Alstrom syndrome in siblings whose initial manifestation was dilated cardiomyopathy.
CASE REPORT: Report of two cases of the syndrome in siblings, whose initial symptomatology was dilated cardiomyopathy, but with different evolutions, with total recovery of cardiomyopathy in one of them. Diagnosis was initially made in the older sister, after seeking Child Neurology service due to horizontal eye movements, previous history of cardiomyopathy, also obesity and hypertriglyceridemia. Her brother also presented dilated cardiomyopathy, but he still has no other symptons of the syndrome. The diagnosis of both was confirmed after molecular analysis.
DISCUSSION: In this report, attention is drawn to the intra-family variability of the disease, both related to the symptoms presented and to the evolution, making it even more difficult to diagnose the syndrome early. This variability suggests that epigenetic factors may play a role in the presentation of the disease, mainly by allelic modification.
Keywords: Cardiomyopathy, dilated. Alstrom syndrome. Siblings.
Introdução
A síndrome de Alstrom (ALMS; OMIM# 203800, http://www.ncbi.nlm.nih.gov/omim) é uma ciliopatia monogênica autossômica recessiva rara, caracterizada por distrofia progressiva de cones e bastonetes, perda auditiva neurossensorial, obesidade infantil e diabetes mellitus tipo 2. A cardiomiopatia dilatada ocorre em aproximadamente 50% dos pacientes durante a infância ou adolescência. Podem ser observadas, ainda, insuficiência renal, disfunção pulmonar, hepática e urológica, com a fibrose sistêmica se desenvolvendo com a idade.1,2
O diagnóstico é difícil, pois as crianças afetadas apresentam um subconjunto de características no início da doença,2 o que torna a prevalência da síndrome também difícil de estimar pela grande probabilidade de os indivíduos com formas atenuadas serem subdiagnosticados.1
Dessa forma, este estudo objetiva relatar dois casos de síndrome de Alstrom em um casal de irmãos, tendo como manifestação inicial a cardiomiopatia dilatada, com a finalidade de demonstrar a variabilidade fenotípica intrafamiliar.
Relato de caso 1
Paciente do sexo feminino atendida em unidade terciária em nível ambulatorial de Neurologia Pediátrica inicialmente aos 2 anos e 10 meses de vida. Apresentava história de cinco meses de evolução de movimento ocular em diversas direções no olho esquerdo, que pioravam ao longo do dia, associado a irritabilidade excessiva e episódios de choro inconsolável.
Primeira filha de pais jovens não consanguíneos. Pré-natal e parto sem intercorrências, peso ao nascer de 3.100g, estatura de 47 cm, perímetro cefálico (PC) de 33 cm. No primeiro mês de vida, diagnosticou-se cardiomiopatia dilatada grave, com indicação de transplante cardíaco. Evoluiu com melhora espontânea. Antecedentes familiares de morte súbita em dois familiares jovens durante a prática de esportes, por parte da família materna, porém sem maiores esclarecimentos.
Ao exame inicial, apresentava peso 17,1 kg (p95), estatura: 94,5 cm (p50), IMC: 19,35 (p99), PC: 46,5 cm (p15). Foram notados movimentos oculares involuntários multidirecionais e hipercinéticos, mais evidentes à esquerda, baixa amplitude, além de hiporreflexia global. Não foram observados sinais de atraso no desenvolvimento.
A investigação complementar com RM de crânio, eletroencefalograma, audiometria, potencial evocado do tronco encefálico (PEATE), cariótipo, exames laboratoriais e tomografia de abdome total com contraste foi normal. O ecocardiograma mostrou função sistólica do VE normal, insuficiência aórtica discreta, conforme demonstrado na tabela 1. A avaliação oftalmológica foi normal, em três momentos diferentes.
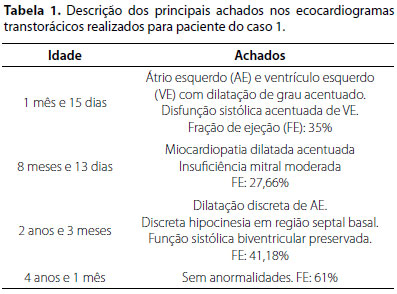
Com os achados clínicos, foi aventada hipótese de síndrome de Alstrom e solicitado teste genético e painel para miocardiopatias, conforme descrito a seguir: - Síndrome de Alstrom (OMIM # 203800) - Gene ALMS1 Posição chr2:73.681.104 variação A > T consequência p.Lys2483* ENST00000613296 cópias Heterozigose (1 cópia) e Gene ALMS1 Posição chr2:73.799.937 variação C > T p.Gln3644* ENST00000613296 Heterozigose (1 cópia).
Paciente segue em seguimento multiprofissional e apresenta, atualmente, obesidade, nistagmo e hipertrigliceridemia. Segue em uso regular de carvedilol.
Relato de caso 2
Durante seguimento do caso anteriormente descrito, a mãe compareceu grávida de 21 semanas. Paciente foi atendido na Neurologia Pediátrica aos cinco meses de vida, devido à história da irmã. Apresentava histórico de nascimento com 40 semanas de idade gestacional, por cesariana devido a líquido meconial e sofrimento fetal agudo, APGAR 9/10, peso 3.564g, estatura 49 cm, PC 34 cm. Gestação não planejada, sem intercorrências. Necessitou de internação aos 43 dias de vida devido a miocardiopatia dilatada.
Realizou sequenciamento NGS do gene ALMS1 com um mês de vida, confirmando-se Heterozigose da variante p.Gln3644* no gene ALMS1: chr2:73.799.937C>T (c.10930C>T; p.Gln3644* - ENST0000000613296). Avaliação oftalmológica normal. Atualmente com miocardiopatia dilatada com disfunção sistólica global do ventrículo esquerdo, insuficiência mitral acentuada, hipertensão pulmonar, forame oval pérvio de 3 mm, FE: 50%, em uso de carvedilol e captopril.
Discussão
Apresentamos dois irmãos com síndrome de Alstrom, cuja apresentação clínica consistiu em miocardiopatia precoce, porém com gravidade diferente. Este foi o segundo caso registrado no Brasil com a síndrome em irmãos, sendo o primeiro em que ambos apresentaram cardiomiopatia como sintoma inicial.3
O diagnóstico clínico da síndrome de Alstrom é baseado em características clínicas cardinais de acordo com as faixas etárias (0 a 2 anos, 3 a 14 anos e acima de 15 anos),2 sendo considerado para o diagnóstico entre 0 a 2 anos, a presença de pelo menos 2 critérios maiores ou 1 maior e 2 menores. São considerados como critérios maiores para essa faixa etária: presença de nistagmo/fotofobia/visão prejudicada; cardiomiopatia e história familiar para síndrome de Alstrom/uma variante patogênica do gene ALMS1. Dentre os critérios menores, estão a obesidade e a perda auditiva neurossensorial.2
Com relação aos sintomas, o probando 1 apresentava cardiomiopatia, obesidade e nistagmo, configurando os critérios mínimos para o diagnóstico da síndrome. Já o probando 2 apresentava cardiomiopatia e história familiar positiva para a síndrome. Importante salientar que o diagnóstico do irmão foi favorecido pelo fato de já apresentar o caso da irmã confirmado.
Interessante ressaltar que, nos dois relatos, o sintoma inicial foi a cardiomiopatia dilatada, a qual apresenta incidência na literatura que varia de 42% a 50%, bem inferior aos demais achados, como as alterações oftalmológicas e auditivas, que variam entre 82 a 100%.2
Nesse sentido, chama a atenção a resolução completa da cardiopatia de um dos irmãos, como também a história evolutiva, que foi totalmente diferente entre eles. Com relação à evolução da cardiomiopatia na síndrome, é sabido que a maioria dos pacientes com miocardiopatia dilatada que sobrevivem apresentam melhora ou resolução da mesma.2 Contudo, olhando exclusivamente para os casos intrafamiliares, observou-se também variabilidade fenotípica, com a maioria evoluindo para melhora, mas outros persistindo com a cardiomiopatia a despeito do tratamento realizado.4-6
Esses achados denotam, assim, a variabilidade fenotípica da síndrome, inclusive do que tange aos sintomas cardíacos. Ttal variabilidade sugere que fatores epigenéticos podem desempenhar um papel na apresentação da doença, principalmente por modificação alélica, como bem demonstrado no relato dos gêmeos homozigóticos que também evoluíram de forma distinta.4,5 Em relação aos irmãos relatados, essa variabilidade também se justifica pela heterogeneidade dos achados genotípicos, uma vez que o probando 1 apresentou duas variantes em heterozigose no mesmo gene, enquanto o probando 2, somente uma.
O artigo em questão relata dois casos de síndrome de Alstrom de ocorrência intrafamiliar, que apesar de terem como manifestação inicial comum a cardiomiopatia infantil, evoluíram de formas distintas entre si. Demonstra, portanto, a variabilidade fenotípica de uma doença rara, atentando também para a dificuldade em se estabelecer o diagnóstico em fases precoces da doença, principalmente antes dos cinco anos de idade, levando muitas vezes à subnotificação.
Referências
1. Marshall AD, Muller J, Collin GB, Milan G, Kingsmore SF, Dinwiddie D et al. Alstrom Syndrome: muttion spectrum of ALMS1. Hum Mutat. 2015 July; 36(7): 660-668.
2. Paisey RB, Barrett T, Carey CM, Hiwot T, Cramb R, White A el tal. Rare disorders presenting in the diabetic clinic: an example using audit of the NSCT adult Alström clinics. Practical Diabetes. 2011;28:340-3.
3. Bahmad F Jr, Costa CSA, Teixeira MS, Barros-Filho J, Viana LM, Marshall J. Familial Alstrom Syndrome: a rare cuse of bilateral progressiva hearing loss. Braz J Otorhinolaryngol. 2014; 80:99-104.
4. Hollander SA, Alsaleh N, Ruzhnikov M, Jensen K, Rosenthal DN, Stevenson D, Manning M. Variable clinical course of identical twin neonates with Alstrom syndrome presentin coincidentally with dilated cardiomyopathy. Am J Med Genet. 2017;9999:1-3.
5. Hoffman JD, Jacobson J, Young TL, Marshall JD, Kaplan P. Familial variable expression of dilated cardiomyopathy in Alstrom syndrome: a report of four sibs. American Journal of Medical Genetics 135A:96-98 (2005).
6. Mahamid J, Lorber A, Horovitz Y, Shalev SA, Collin GB, Naggert JK et al. Extreme clinical variability of dilated cardiomyopathy in two siblings with Alstrom syndrome. Pediatr Cardiol. 2013 February; 34(2): 455-458.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()