Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 22(4) - Dezembro 2022
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Pseudoacondroplasia: relato de uma causa incomum de baixa estatura
Pseudoachodroplasia: case report of an unusual cause of short stature
Suely Keiko Kohara1; Patricia Tessari2; Maria Luíza Floriano2
DOI:10.31365/issn.2595-1769.v22i4p163-165
1. Universidade da Região de Joinville - UNIVILLE, Medicina, Pediatria - Joinville - Santa Catarina - Brasil
2. Universidade da Região de Joinville - Univille, Medicina - Joinville - Santa Catarina - Brasil
Endereço para correspondência:
marialuizafloriano@hotmail.com
Recebido em: 20/07/2021
Aprovado em: 29/11/2022
Resumo
INTRODUÇÃO: A pseudoacondroplasia é a segunda forma mais comum de displasia esquelética. É uma condição caracterizada pelas manifestações diversas no esqueleto axial e apendicular. Objetivo: Descrever um paciente com pseudoacondroplasia e relembrar aos pediatras desse diagnóstico diferencial na investigação de baixa estatura.
DESCRIÇÃO DO CASO: Paciente masculino de 5 anos e 7 meses com baixa estatura, claudicação, encurtamento de mãos e genu valgo à esquerda. As alterações radiográficas e análise genética confirmaram o diagnóstico de pseudoacondroplasia.
DISCUSSÃO: A pseudoacondroplasia é uma displasia esquelética caracterizada por baixa estatura desproporcional e osteoartrite de início precoce. Esta condição é causada por uma mutação na proteína oligomérica da matriz da cartilagem (COMP), expressa principalmente nos condrócitos, causando morte prematura destas células do tecido cartilaginoso durante o crescimento ósseo, o que leva a alterações articulares diversas. Apesar de rara, é a segunda forma mais comum de displasia esquelética e deve ser lembrada na investigação de crianças com distúrbios do crescimento.
Palavras-chave: Acondroplasia. Estatura. Insuficiência de crescimento.
Abstract
INTRODUCTION: Pseudochondroplasia is the second most common form of skeletal dysplasia. It is a condition characterized by diverse manifestations in the axial and appendicular skeleton.
OBJECTIVE: To describe a patient with pseudoachondroplasia and remind pediatricians of this differential diagnosis in the investigation of short stature. Case description: A 5 years and 7 months old male patient presented with short stature, lameness, shortened hands and left genu valgus. Radiographic findings, and genetic analysis confirmed the diagnosis of pseudoachondroplasia.
DISCUSSION: Pseudochondroplasia is a skeletal dysplasia characterized by disproportionate short stature and early-onset osteoarthritis. This condition is caused by a mutation in the cartilage oligomeric matrix protein (COMP), expressed mainly in chondrocytes, that causes premature death of these cartilage tissue cells during bone growth, which leads to several joint changes. Although rare, it is the second most common form of skeletal dysplasia and should be considered when investigating children with growth disorders.
Keywords: Achondroplasia. Body Height. Failure to Thrive.
INTRODUÇÃO
A pseudoacondroplasia foi descrita pela primeira vez em 1959 por Maroteaux e Lamy e tem prevalência aproximada de 1/30.000, sendo a segunda forma mais comum de displasia esquelética.1,2 É uma condição de herança autossômica dominante que se caracteriza pelas alterações no esqueleto axial e apendicular, com manifestações clínicas como baixa estatura, escoliose, frouxidão ligamentar e encurtamento das mãos, características que são reconhecidas, geralmente, a partir dos dois anos de idade.1,3,4
Este artigo foi submetido ao Comitê de Ética em Pesquisa e aprovado sob parecer nº 47057921.7.3001.5362 e tem como objetivo a descrição de um paciente com pseudoacondroplasia, a fim de relembrar aos pediatras acerca deste diagnóstico diferencial na investigação de baixa estatura.
DESCRIÇÃO DO CASO
D.R.A, masculino, 6 anos e 3 meses, atendido em serviço de endocrinologia pediátrica por atraso de crescimento. O peso de nascimento era 3.000g e comprimento de 49cm. Sem história de doenças pregressas ou cirurgias. A estatura da mãe era 154 cm e do pai, 163 cm.
Ao exame, apresentava estatura de 102 cm (5 cm abaixo do 3° percentil), peso de 15,4 kg (1,5kg abaixo do 3° percentil), envergadura de 92cm e segmento inferior de 43cm (relação SS/SI= 1,37), pré-púbere, mãos curtas, genu valgo à esquerda e marcha claudicante.
Os exames laboratoriais não mostravam alterações: glicemia de jejum: 85mg/dL, hemograma normal, cálcio: 9,8mg/dL, fósforo: 6,12mg/dL, fosfatase alcalina: 138U/L, magnésio: 2,3 mg/dL, sódio: 130mEq/L, potássio: 4,4mEq/L, anticorpo anti-transglutaminase tecidual (IgA): negativo, TSH: 3,8UI/ml, T4L: 1,02ng/dL, IGF-1: 114 ng/ml (VR 50 a 286), cortisol: 8,47ug/dL, PTH 29 pg/ml, calciúria: 10mg/24h, fosfatúria: 264mg/24h. A velocidade de crescimento era de 6,5cm/a. A radiografia de mãos realizada aos 5 anos e 6 meses evidenciava uma idade óssea de 2 anos e 8 meses, e mostrava encurtamento dos metacarpos com irregularidade cortical das bases, irregularidade cortical das epífises distais de rádio e ulna (demonstrado na Figura 1). O caso foi enviado ao site de auxílio diagnóstico das osteocondrodisplasias (www.odc.med.br), que sugeriu o diagnóstico de pseudoacondroplasia. Foi solicitada análise genética (painel NGS/exoma TW), a qual identificou uma variante em heterozigose no gene COMP, associada a pseudoacondroplasia e displasia epifisária múltipla. Essa variante missense c.2156G>A consiste na substituição de um nucleotídeo (G>A) na posição 2156 no exon 18, promovendo a troca de uma glicina no códon 719 por um ácido aspártico (p.(Gly719Asp)).
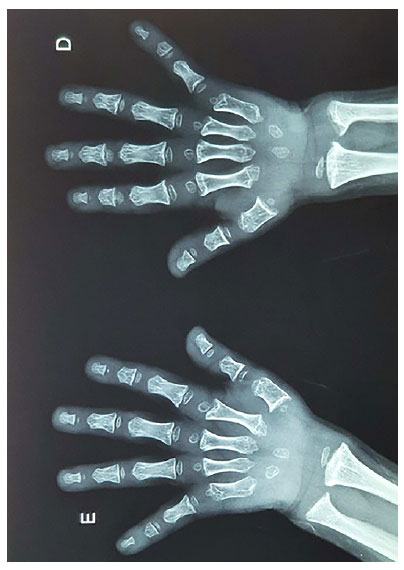
Figura 1. Radiografia de mãos (encurtamento dos metacarpos com irregularidade cortical das bases, irregularidade cortical das epífises distais de rádio e ulna).
Portanto, foi confirmado o diagnóstico de pseudoacondroplasia e o paciente foi encaminhado para avaliação com neuropediatra e ortopedista, além de manter acompanhamento com endocrinopediatra.
DISCUSSÃO
A base fisiopatológica da pseudoacondroplasia está na displasia de epífises e metáfises dos ossos longos, ocasionada primariamente por uma mutação no gene codificador da proteína oligomérica da matriz da cartilagem (COMP).1 A mutação deste gene da COMP torna a cartilagem articular incapaz de suportar cargas cotidianas, acarretando deformidades em todas as áreas do esqueleto axial e apendicular, sobretudo em membros inferiores.1
Clinicamente, esses pacientes têm peso e comprimento normais ao nascer e desenvolvem baixa estatura desproporcional, com encurtamento dos membros em relação ao tronco, alterações de marcha, joelhos proeminentes e cifoescoliose.4 Também, esta condição se associa a quadros de artralgia ao longo da vida e à osteoartrite de início precoce, sendo a dor crônica a complicação mais debilitante da doença, uma vez que compromete a mobilidade, realização de atividade física e a qualidade de vida.5,6 Outras características incluem marcha anserina, amplitude limitada de movimento nos cotovelos e quadris, braquidactilia e curvaturas anormais da coluna em alguns indivíduos. Já as características faciais, o tamanho da cabeça e inteligência estão preservados.7 Ainda, a subossificação do processo odontoide pode levar à instabilidade da parte superior da coluna, o que requer intervenção cirúrgica.8
O diagnóstico é baseado em achados clínicos e radiográficos característicos.9 Nos exames radiográficos, alguns achados como o achatamento dos corpos vertebrais, a braquidactilia e as alterações das metáfises e epífises são característicos.4 Todos os ossos longos das mãos são encurtados, os ossos do carpo tornam-se irregulares e a maturação óssea é retardada.8 O tratamento envolve acompanhamento ortopédico e controle da dor, devido à deterioração das articulações.4 Também fisioterapia, tratamento das deformidades da coluna e correções cirúrgicas fazem parte da terapia, devendo a cirurgia ser postergada até o final do período de crescimento.10
Conclui-se que o paciente descrito apresentava baixa estatura, genu valgo, claudicação e encurtamento dos membros, além de idade óssea atrasada. Em conjunto com história clínica e imagens radiológicas, o diagnóstico de pseudoacondroplasia foi suspeitado, sendo confirmado pelo teste genético mostrando mutação no gene COMP. Apesar de rara, essa patologia deve ser lembrada, principalmente após os dois anos de vida, nos quadros de insuficiência de crescimento associado a alterações articulares.
REFERÊNCIAS
1. Weiner DS, Guirguis J, Makowski M, Testa S, Shauver L, Morgan D. Orthopaedic manifestations of pseudoachondroplasia. J Child Orthop. 2019;13(4):409-416. doi:10.1302/1863-2548.13.190066
2. Maroteaux P, Lamy M. Les formes pseudo-achondroplasiques des dysplasies spondylo-epiphysaires [Pseudo-achondroplastic forms of spondylo-epiphyseal dysplasias]. Presse Med. 1959 Feb 25;67(10):383-6
3. Briggs MD, Hoffman SM, King LM, Olsen AS, Mohrenweiser H, Leroy JG, et al. Pseudoachondroplasia and multiple epiphyseal dysplasia due to mutations in the cartilage oligomeric matrix protein gene. Nat Genet. 1995 Jul;10(3):330-6. doi: 10.1038/ng0795-330.
4. Bacino CA. Skeletal dysplasias: Specific disorders. UpToDate. 2020. Disponível em: <http://www.uptodate.com/online>. Acesso em: 17/02/2021
5. Posey, K. and Hecht, J. Novel therapeutic interventions for pseudoachondroplasia. Bone. 2017 Sep. 102, pp.60-68. doi: https://doi.org/10.1016/j.bone.2017.03.045
6. Gamble C, Nguyen J, Hashmi SS, Hecht JT. Pseudoachondroplasia and painful sequelae. Am J Med Genet A. 2015 Nov;167A(11):2618-22. Epub 2015 Jul. doi: 10.1002 / ajmg.a.37253.
7. Genetic and Rare Diseases Information Center. EUA. Pseudoachondroplasia. https://rarediseases.info.nih.gov/diseases/4540/pseudoachondroplasia.
8. Posey KL, Alcorn JL, Hecht JT. Pseudoachondroplasia/COMP - translating from the bench to the bedside. Matrix Biol. 2014 Jul;37:167-73. doi: 10.1016/j.matbio.2014.05.006.
9. Unger S, Hecht JT. Pseudoachondroplasia and multiple epiphyseal dysplasia: New etiologic developments. Am J Med Genet. 2001 Winter;106(4):244-50. PMID: 11891674.
10. Radlovic V, Smoljanic Z, Radlovic N, Jakovljevic M, Lekovic Z, Ducic S, et al. Srp Arh Celok Lek. 2013 Sep-Oct;141(9-10):676-679.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()