Revista de Pediatria SOPERJ
ISSN 1676-1014 | e-ISSN 2595-1769
Número atual: 20(3) - Setembro 2020
- Imprimir
- Indicar
- Estatísticas
- (0)
Comentários - Como Citar
- Download da Citação
- Artigos Relacionados
-
Outros dos
Autores
Relato de Caso
Displasia ectodérmica hipoidrótica em paciente do sexo feminino: um relato de caso
Hypohidrotic ectodermal dysplasia in a female patient: a case report
Marcos Antonio Coutinho Costa Rodrigues; Stephany Pina Da Cunha Nascimento Mesquita; Anne Karoline Tomé Briglia; Cássia Iasmin De Souza Nascimento; Charlote Aguiar Buffi Briglia; Ellem Tatiani De Souza Weimann
DOI:10.31365/issn.2595-1769. v20i3p102-105
Universidade Federal De Roraima, Curso De Medicina - Boa Vista - Roraima - Brasil
Endereço para correspondência:
Recebido em: 17/04/2020
Aprovado em: 20/05/2020
Instituição: Universidade Federal De Roraima, Curso De Medicina - Boa Vista - Roraima - Brasil
Resumo
INTRODUÇÃO: A displasia ectodérmica hipoidrótica ou anidrótica (DEHA) é uma genodermatose, de epônimo síndrome de Christ-Siemens-Touraine, com desenvolvimento de anormalidades estruturais e funcionais de dois ou mais apêndices ectodérmicos.
OBJETIVO: Relatar um raro caso clínico de displasia ectodérmica hipoidrótica (DEH), doença de herança ligada ao X, em uma criança do sexo feminino.
DESCRIÇÃO DO CASO: A paciente feminina nasceu prematura, apresentando atrasos leves do desenvolvimento neuropsicomotor relacionados a intercorrências em período neonatal, apresentando fácies sindrômicas, rarefação de pelos e fâneros e intolerância ao calor.
DISCUSSÃO: A DEHA é uma genodermatose rara, e a tríade clínica de hipotricose, dentição anômala e hipoidrose fecha diagnóstico da paciente supracitada para DEHA, importante para o conhecimento do pediatra generalista.
Palavras-chave: Displasia ectodérmica; Síndrome de Christ-Siemens-Touraine; Dermatopatias genéticas.
Abstract
INTRODUCTION: Hypohydrotic or anhydrotic ectodermal dysplasia (DEHA) is a genodermatosis, of the eponymous Christ-Siemens-Touraine syndrome, with the development of structural and functional abnormalities of two or more ectodermal appendages.
OBJECTIVE: To report a rare clinical case of hypohidrotic ectodermal dysplasia (DEH), an X-linked inheritance disease, in a female child.
CASE DESCRIPTION: The female patient was born premature, with mild delays in neuropsychomotor development related to complications in the neonatal period, presenting syndromic facies, thinning of hair and sex and heat intolerance.
DISCUSSION: DEHA is a rare genodermatosis and the clinical triad of hypotrichosis, anomalous dentition and hypohidrosis closes the diagnosis of the aforementioned patient for DEHA, which is important for the knowledge of the general pediatrician.
Keywords: Ectodermal dysplasia; Genetic Diseases; Inborn; Ectodermal Dysplasia; Hypohidrotic; Autosomal Recessive.
INTRODUÇÃO
A displasia ectodérmica hipoidrótica ou anidrótica (DEHA), ou ainda síndrome de Christ-Siemens-Touraine, é uma genodermatose com desenvolvimento de anormalidades estruturais e funcionais de dois ou mais apêndices ectodérmicos.1 A DEHA tem herança ligada ao X, com incidência de aproximadamente 1:100.000 nascidos, sendo que 90% dos pacientes são do sexo masculino. Segundo a literatura, a síndrome completa raramente ocorre em mulheres, podendo as mães dessas crianças ser portadoras do gene casual e terem manifestações discretas.2
As alterações clínicas compreendem: sudorese ausente (anidrose) ou reduzida (hipoidrose), hipotricose, anodontia total ou parcial. Na apresentação completa da síndrome, podem-se encontrar: rugas frontais, nariz em sela, lábios espessos evertidos, orelhas grandes, tíbia proeminente e pele seca. A alopecia é uma das primeiras características que chamam a atenção, mas raramente é total. Os cabelos são esparsos, finos, secos, permanentemente curtos e pouco pigmentados. As sobrancelhas são esparsas ou ausentes, mas os cílios são normais, em geral. Os pelos corporais podem estar ausentes ou esparsos. Os dentes incisivos e/ou caninos são caracteristicamente cônicos e pontiagudos e são a chave para o diagnóstico da síndrome. Anormalidade ungueal ocorre em 50% dos casos e se caracteriza por unhas finas e enrugadas.3
Em virtude da significativa raridade do quadro clínico em meninas, a presente pesquisa tem como objetivo relatar um caso clínico raro, para conhecimento da comunidade científica, da displasia ectodérmica hipoidrótica (DEH) em uma criança do sexo feminino. O caso foi identificado no ambulatório de neuropediatria e encaminhado para dermatopediatria, tornando-se interessante para relato pelas manifestações clínicas características para diagnóstico de DEH em lactente do sexo feminino. Faz-se necessário que pediatras e demais profissionais da área da saúde tenham conhecimento a respeito deste quadro clínico, visto que uma boa anamnese e exame físico direcionado auxiliam no diagnóstico clínico de DEH e numa terapêutica eficaz.
DESCRIÇÃO DO CASO
Paciente do sexo feminino, natural de Boa Vista-RO, nasceu com 32 semanas, via cesárea, com peso ao nascer de 1.214 gramas, estatura de 34 cm e perímetro cefálico de 27 cm. Genitora (G3P2cA1) teve 13 consultas pré-natais, sorologias negativas, evoluiu com intercorrências - diabetes gestacional e doença hipertensiva gestacional - necessitando de interrupção da gestação devido a pressão arterial em 230 x 110 mmHg e sofrimento fetal. Nos antecedentes familiares, os pais, ambos com 27 anos, não são consanguíneos, sem comorbidades e sem história familiar (genitores) de doenças autoimunes e genéticas. Irmão sem comorbidades.
Após nascimento, a paciente ficou dois meses internada em Unidade de Terapia Intensiva Neonatal (UTIN), devido ao desconforto respiratório precoce, evoluiu com instabilidade hemodinâmica devido a hipertensão pulmonar moderada, identificada em ccocardiograma, necessitando de ventilação pulmonar mecânica, HOOD e O2 circulante, feito uso de sildenafila para estabilização de quadro clínico.
Aos cinco meses, iniciou acompanhamento no ambulatório de neuropediatria, por apresentar atraso do desenvolvimento neuropsicomotor (DNPM), caracterizado por sustento cefálico incompleto, postura assimétrica, com sinais precoces de hipertonia de membros, polegares em adução, reflexos osteotendíneos vivos, sem clônus; além disso, apresentava fácies sindrômica com rarefação de fâneros. Foi solicitado cariótipo (46XX), sendo então encaminhada para avaliação dermatológica.
No 8º mês de vida, em consulta na dermatopediatria, genitora relatou intolerância ao calor e ao exame físico, identificando-se: rarefação capilar em sobrancelha e cílios, haste pilosa normal, nariz em sela, discromia em ponta nasal, unhas sem alterações, sem dentição. Com hipótese de genodermatose, foi indicado seguimento clínico.
Aos 10 meses permaneceu sem dentição, com intolerância ao calor e sem sudorese, apresentando aumento de densidade capilar em couro cabeludo, presença de um terço das sobrancelhas e cílios, xerodermia ictiosiforme em pernas. Aos 14 meses, evoluiu com aumento de densidade capilar, presença completa de cílios, pápulas hipocrômicas em ponta nasal com vesículas lunares hipercrômicas. Com 19 meses, continuou com atraso motor leve, início de fala em pequenas palavras, deambulação com base alargada, presença de discreta umidade em região poplítea, nascimento de dentes incisivos centrais cônicos, cílios completos e aumento de pelos, fechando diagnóstico clínico para DEH (Figura 1 e Figura 2).
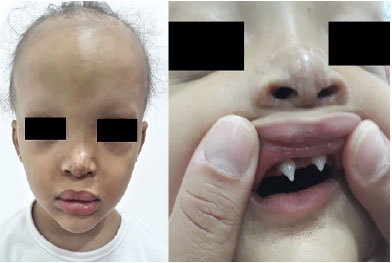
FIGURA 1 . Paciente com rarefação capilar e de sobrancelhas, nariz em sela com discromia em ponta nasal, lábios evertidos e dentes incisivos cônicos pontiagudos.
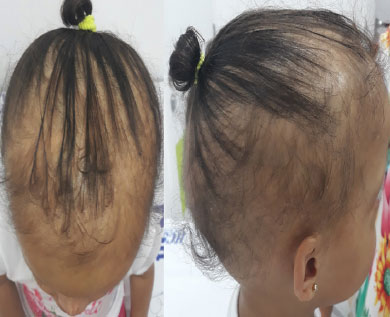
FIGURA 2 . Paciente com hipotricose: cabelos esparsos, finos e curtos.
Não se realizou exame molecular, nem biópsia de pele, uma vez que a criança apresentava fenotipia característica para DEH. Considerando que a biópsia seria para classificação, não modificando a conduta, e que para a realização do procedimento seria necessário sedação da paciente, ao avaliar riscos x benefícios, a equipe optou por não realizar o procedimento.
DISCUSSÃO
A displasia ectodérmica (DE) envolve um conjunto heterogêneo de alterações, abrangendo os tecidos derivados do ectoderma.4 Existem diferentes tipos de DE, representando um complexo grupo de doenças com mais de 170 características clínicas.5,6,7 Desde 1984, são descritas e classificadas mais de 117 formas de DE baseadas em combinações clínicas específicas e características morfológicas.8,9 A forma mais comum de DE é a recessiva ligada ao cromossomo X, a hipoidrótica ou anidrótica.10 Ou, mais raramente, mutações em genes autossômicos dominantes e recessivos.6,7,11 Por este motivo, apenas homens expressam de forma completa suas características em relatos de literatura.11
As formas mais comuns de DE caracterizam-se pela ausência ou defeito nos dentes, pelos, pele, unhas, glândulas salivares e glândulas sudoríparas.9 Porém, as DE revelam sinais de múltiplas origens embrionárias.8 O nome original permanece porque os defeitos ectodérmicos são mais severos ou mais evidentes que os demais.5
Em relação aos diagnósticos diferenciais principais, temos a DEH ou síndrome de Clouston, de herança autossômica dominante, caracterizada pela tríade: alopecia, unhas distróficas e ceratodermiapalmoplantar, além de cabelos espaços, finos, claros e de crescimento lento, tendo como principais características que a diferenciam: os dentes normais, sendo comuns as cáries dentárias, e a sudorese não alterada (hidrose). A outra patologia é a síndrome displásica ectodérmica, que associa as alterações clássicas das doenças ectodérmicas à fragilidade cutânea e à ceratodermiapalmoplantar progressiva e grave.2
Em torno de 94% dos casos de DEHA ocorrem devido à mutação do gene ED1, em Xq12-q13. São descritas cerca de 53 diferentes mutações no gene ED1.7,11 O gene ED1 é responsável por codificar a ectodisplasina, uma proteína de colágeno transmembrana semelhante a proteínas que ligam o fator de necrose tumoral. A ectodisplasina possui duas formas, a ED1-A1 e a EDA-A2, as quais se ligam, respectivamente, aos receptores EDAR e XEDAR. Ao ocorrer tais ligações, são ativadas as cascatas do fator nuclear (NF)êB e do JNK/cfos/c-jun, ambas responsáveis pela ativação de sinais para atuação do fator de crescimento da epiderme, que permitem a diferenciação da epiderme e de seus anexos.6,7,11 Desta maneira, conclui-se que a ectodisplasina tem função na comunicação, sobrevivência e crescimento celular.6
As manifestações da DEHA envolvem redução ou ausência das glândulas écrinas, sebáceas e lacrimais.11 Por isso, sinais como tricodisplasia, defeitos dentários, onicodisplasia ou disidrose, além de outro sinal afetando uma estrutura de origem ectodérmica, estão presentes.8
A alteração glandular manifesta-se com a impossibilidade de suar; por tal motivo, ocorre elevação da temperatura corporal, que complica com hipertermia e convulsões febris. A redução ou ausência de glândulas lacrimais leva a dacriocistites de repetição. A pele pode manifestar xerodermia, placas de liquenificação e ser sede de dermatites atópicas. O cabelo é fino, seco e hipocrômico; além disso, pode ocorrer alopecia. Quanto às anomalias ungueais, pode haver unhas hiperconvexas ou hipertróficas, distróficas ou queratinizadas, ou ausentes. Os dentes são de tamanho pequeno e de forma cônica.12 Também se encontra hipodontia ou adontia. As anomalias faciais demonstram fácies características com alterações como fronte proeminente, pigmentação periorbitária, orelhas pontiagudas de baixa implantação, nariz em sela e protrusão de lábio inferior.13
A suspeita diagnóstica deve se iniciar em crianças pequenas com inexplicável febre duradoura e anidrose. Em crianças mais velhas, a suspeita diagnóstica, quando se manifestam as características clínicas completas (hipotricose, dentição anômala e hipoidrose), não é difícil de ser feita. A biópsia da pele confirma a classificação.4
O tratamento é sintomatológico. As medidas terapêuticas se concentram no controle da temperatura do paciente, uso de emolientes para a pele e supervisionar a dentição. A terapia corretiva dentária, com a utilização de prótese, auxilia o paciente na nutrição e na melhora da aparência. Em adolescente e adultos, a cirurgia plástica pode ser realizada para corrigir deformidades.14,4 É recomendado que um grupo composto por geneticista, dermatologista, otorrinolaringologista, pediatra, odontopediatra, protesista, fonoaudiólogo e psicólogo acompanhe o paciente.9
Ressalta-se que as intercorrências ocorridas em período neonatal explicam o atraso motor leve que se resolveu com o tempo; logo, não é possível relacionar o atraso do DNPM da paciente com a DEHA, uma vez que houve internação em UTIN com quadro clínico de instabilidade hemodinâmica.
O conhecimento do quadro clínico relatado é de grande importância para o diagnóstico diferencial de genodermatoses, visto que o caso clínico é de uma síndrome rara, sobretudo na forma completa em pacientes do sexo feminino, com diagnóstico a depender da evolução clínica. Atualmente, a paciente descrita encontra-se estável e em acompanhamento por equipe multidisciplinar com neuropediatra, dermatologista, odontopediatra e fisioterapeuta.
REFERÊNCIAS
1. Ferreira CS, Ferreira RAMH, Fernandes MLMF, Branco KMGR, Arantes RR, Leão LL. Displasia ectodérmica: relato de caso. Arq Odontol. 2012; 48(1):47-52.
2. Azulay RD. Dermatologia. 6ª ed. Rio de Janeiro: Guanabara Koogan; 2015.
3. Succi IB, Fontenelle E. Caso para diagnóstico. Displasia ectodérmica: síndrome de Christ-Siemens-Touraine. Na Bras Dermatol. 2009; 84(2):194-196.
4. Coskun Y, Bayraktaroglu Z. Pathological case ofthemonth. Archives of Pediatrics and Adolescent Medicine. 1997; 151(7):741-742.
5. Sarmento VA, et al. Displasia ectodérmica: revisão da literatura e relato de casos clínicos. Sitientibus. 2006; (34):87-100.
6. Lamartine J. Toward a new classificationofectodermaldysplasias. Clinical and Experimental Dermatology. 2003; 28:351-355.
7. Priolo M, Silengo M, Lerone M, Ravazzolo R. Ectodermal Dysplasias: Notonlya 'skin' deep. Clinical Genetics. 2000; 58(6):415-430.
8. Freire-Maia N, Pinheira M. Ectodermal dysplasias: a clinical and genetic study. American Journal of Medical Genetics. 1984; 28(4):1025-1026.
9. Bakri H. Clinical management ofectodermaldysplasia. Journal of Clinical Pediatric Dentistry. 1995; 19(3):167-172.
10. Murdoch-Kinch C, Miles DA, Poon CK. Hypodontia and Nail Dysplasia Syndrome: Report of a Case. Oral Surgery, Oral Medicine, Oral Pathology. 1993; 75(3):403-406.
11. Gun NY, et al. Mutation in the ED1 Gene, Ala349Thr, in a Korean Patient with X-linked hypohidrotic ectodermal dysplasia. Pediatric Dermatology. 2004; 21(5):568-572.
12. Garcia JG, Ruiz GH, Borges FM. Manifestaciones ORL de la displasia ectodérmica hipo-hidrótica. Arquivos Internacionais de Otorrinolaringologia. 2005; 55:1-6.
13. Koerner HN, Bettega S, Mocellin M. Rinite atrófica: relato de caso associado a displasia ectodérmica. Arquivos Internacionais de Otorrinolaringologia. 2006; 10:1-6.
14. Paller AS. Hereditary disease of skin, hair, nails, and skin structure: In: Maldonado L, Parish B, eds. Pediatric dermatology. 1989; 85.
![]()
![]()
![]()
![]()

Copyright 2025: SOPERJ | Rua da Assembleia, 10, sala 1.812, Rio de Janeiro RJ | Whatsapp: (21) 2531-3313 / tel: (21) 3923-5234 ![]()